











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
La vena cava superior izquierda persistente (VCSIP) es una anomalía congénita del desarrollo venoso sistémico poco frecuente, presente en el 0,3% al 0,5% de la población general, y en el 10%-12% de pacientes con cardiopatías congénitas (CC)1-8.
Cuando se asocia a ausencia de la vena cava superior derecha (VCSD) su incidencia es aún menor, estando presente en el 0,09%-0,13% de pacientes con CC2,5-8.
Asimismo, la incidencia de la VCSIP es superior en pacientes con alteraciones cromosómicas (7,8%)1,2,6.
En la gran mayoría de los casos, la VCSIP drena hacia la aurícula derecha a través de un seno coronario (SC) dilatado y sólo en raras ocasiones ingresa directamente en la aurícula izquierda (8%) entre las venas pulmonares y el apéndice izquierdo, lo que resulta en un retorno venoso sistémico parcial anómalo2,3,8,9. Esta situación también puede generar alteraciones del sistema de conducción (fibrilación auricular o ventricular) o favorecer el desarrollo de alguna cardiopatía congénita como lesiones obstructivas del tracto de salida izquierdo (coartación de aorta)9.
La dilatación del seno coronario es el principal signo ecocardiográfico de sospecha. Lo más frecuente es que se trate de un diagnóstico casual aislado, sin embargo, es posible su diagnóstico durante el período fetal siendo importante hacer un diagnóstico diferencial con otras CC que asocien también dilatación del seno coronario tales como la comunicación interauricular ostium primum, la atresia mitral y el retorno venoso anómalo total a seno coronario.
Existen muy pocas series de casos descritos en la literatura por lo que presentamos dos casos recientemente diagnosticados en nuestro hospital durante el período fetal y confirmados posnatalmente1-6.
Ambos fueron embarazos controlados y con buena evolución clínica tras un seguimiento estrecho que nacieron a término mediante parto eutócico y con peso adecuado para la edad gestacional.
Casos clínicos
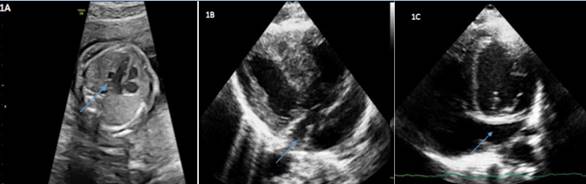
El primer caso es diagnosticado en la semana 30, tras sospecharse al visualizar un seno coronario dilatado (Figura 1A) con una imagen patológica en el corte de los tres vasos, un vaso a la izquierda del ductus en el plano tres vasos tráquea (3VT) que parecía drenar en el mismo, con un dudoso vaso a la derecha de la aorta, no claramente captante en el Doppler. No se observan otras alteraciones anatómicas en otros órganos.
Como antecedentes familiares, se trata de una madre sana de 25 años (segunda gestación) sin antecedentes personales. No hay antecedentes en la familia de cardiopatías congénitas ni de cromosomopatías ni de enfermedades inmunológicas. No hay consanguinidad entre los padres. El embarazo es normoevolutivo y controlado sin incidencias. Las serologías fueron negativas y como único antecedente infeccioso presenta un estreptococo del grupo B (SGB) positivo que se trata con profilaxis completa. Nace tras un parto eutócico a la semana 40, sin incidencias. Apgar 10/10 sin precisar reanimación neonatal.
Tras el nacimiento se realiza cribado de CC mediante pulsioximetría en las primeras 24 horas de vida en la planta de maternidad que supera. En la primera exploración al nacimiento no se observan rasgos dismórficos y la auscultación cardiopulmonar es normal. A las 48 horas de vida, previo al alta de la maternidad, se solicita interconsulta a cardiología infantil ante los hallazgos prenatales, realizándose ecocardiograma donde se confirma VCSIP junto con agenesia VCSD observándose también posnatalmente una dilatación del seno coronario (Figura 1B y 1C). Así mismo se observa una CIA tipo OS pequeña junto con una válvula mitral de tamaño normal, pero con apertura algo restringida por un velo posterior corto. También se realiza electrocardiograma (ECG) con resultado normal. No precisa otras pruebas complementarias en su primera valoración por cardiología infantil.
Posteriormente se realiza seguimiento en consultas externas, donde se confirma evolutivamente cierre de la CIA OS y normalización del aspecto de los velos de la válvula mitral siendo dada de alta a los dos años de seguimiento. No precisa tratamiento desde el punto de vista cardiológico en ningún momento.
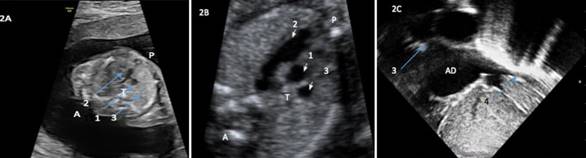
El segundo caso es diagnosticado en la semana 20 tras visualizarse en el corte 3VT un vaso a la izquierda de la arteria pulmonar que correspondía a la vena cava superior izquierda sin visualizarse vena cava superior derecha, junto dilatación del seno coronario (Figura 2A), Nótese la diferencia del plano 3VT en un paciente sin esta anomalía (Figura 2B). No se observan otras alteraciones anatómicas en otros órganos.
Como antecedentes familiares, se trata de una madre de 32 años (primera gestación) que como antecedentes personales presenta una hiperprolactinemia no tratada y que ha sido intervenida quirúrgicamente a nivel maxilofacial en varias ocasiones, sin relevancia para el caso. No hay antecedentes en la familia de cardiopatías congénitas ni de cromosomopatías ni de enfermedades inmunológicas. No hay consanguinidad entre los padres.
El embarazo es normoevolutivo y controlado sin incidencias, salvo un test O’Sullivan precoz patológico con sobrecarga oral de glucosa posterior normal. Las serologías fueron negativas y no tiene otros antecedentes infecciosos. Nace tras un parto eutócico a la semana 41, sin incidencias. Apgar 10/10 sin precisar reanimación neonatal.
Tras el nacimiento se realiza cribado de CC por pulsioximetría en las primeras 24 horas de vida en la planta de maternidad que supera. En la primera exploración al nacimiento no se observan rasgos dismórficos y la auscultación cardiopulmonar es normal. A las 48 horas de vida, al igual que al paciente anterior, se solicita interconsulta a cardiología infantil ante los hallazgos prenatales, realizándose ecocardiograma que confirma el diagnóstico fetal (se visualiza la entrada de la VCI en la AD y la ausencia de VCS) sin objetivarse cardiopatía estructural asociada (Figura 2C). Se realiza ECG basal con resultado normal. No precisa otras pruebas complementarias en su primera valoración por cardiología infantil, siendo dado de alta de la consulta tras esta primera evaluación.
Discusión
Como ya se ha comentado, el desarrollo de la VCSIP junto con la ausencia de VCSD se produce tras la obliteración durante la embriogénesis de la vena cardinal anterior derecha con persistencia de la vena cardinal anterior izquierda que drena en la aurícula derecha (AD) a través del seno coronario1-10.
En el 80%-90% de los casos, la VCPSI coexiste con VCSD, por lo que tendremos un doble sistema de vena cava superior6-8. En estos casos la VCSIP habitualmente drena hacia la aurícula derecha a través de un seno coronario dilatado, sin cursar manifestaciones clínicas secundarias a esta anomalía. No obstante, en el 10%-20% de los casos puede drenar directamente en la aurícula izquierda por medio de un seno coronario sin techo, o a través de la vena pulmonar superior izquierda, lo que resulta en un retorno venoso sistémico parcial anómalo.
Así mismo, el mecanismo que provoca la persistencia de la VCSI además de generar la ausencia de la VCSD, puede desarrollar de manera concomitante alguna anomalía cardiaca congénita (lesiones obstructivas del tracto de salida izquierdo) o alteraciones del desarrollo del sistema de conducción2,9.
En relación a las alteraciones del sistema de conducción, se han descrito alteraciones en el desarrollo o hipoplasia del nódulo sinusal que pueden provocar disfunción del mismo, ritmo auricular no sinusal, además de alteraciones en el nodo aurículoventricular, facilitando la posibilidad de bloqueo auriculoventricular2,5. De igual manera, se han descrito taquiarritmias tales como fibrilación auricular o ventricular debido a la dilatación del seno coronario, que puede producir un estrechamiento del nodo AV o el haz de His8,9.
En cuanto a las cardiopatías congénitas asociadas, se han descrito defectos del septo auricular e interventricular, defectos atrioventriculares, lesiones obstructivas izquierdas como la coartación de la aorta, cor triatriaum y anomalías conotruncales como la tetralogía de Fallot y el ventrículo derecho de doble salida, dependiendo de las series esta incidencia puede ser hasta del 80%2-9.
Se ha comentado también que la VCSIP junto con agenesia de VCSD puede estar presente en diferentes cromosomopatías o síndromes siendo las más frecuentemente descritas la trisomía del 18, la del 21 y el síndrome VACTERL1,2,6,7.
De igual manera se han descrito anomalías extracardíacas sin relación con síndromes, siendo la más frecuentemente asociada la atresia esofágica, aunque también se han observado otras asociaciones mayores como la hernia diafragmática o menores como la hipoplasia del timo o arteria umbilical única1-7.
Durante el examen fetal rutinario en la semana 20 del embarazo, la VCSIP puede diagnosticarse mediante el corte 3VT con un vaso supernumerario a la izquierda de la arteria pulmonar y el ductus arterioso o mediante hallazgos indirectos, como un seno coronario dilatado en el corte de cuatro cámaras. Si se asocia a ausencia de la VCSD, en vez de visualizarse cuatros vasos en un corte transverso se visualizan tres, pero con una disposición anormal de derecha a izquierda: aorta, arteria pulmonar y VCSIP.
Ante el hallazgo de esta anomalía del sistema venoso sistémico en la época fetal cabe destacar la importancia por un lado de su correcta descripción durante este periodo junto con la exclusión de otras alteraciones anatómicas.
Al igual que en la literatura revisada, nuestros dos casos fueron diagnosticados durante la ecografía del segundo trimestre, ante la realización de uno de los planos rutinarios y estandarizados (3VT).
A pesar de la sospecha diagnóstica de esta anomalía congénita del desarrollo venoso sistémico, en ambos casos se descartaron otras anomalías asociadas o cardiopatías debido al resto de la normalidad de las imágenes fetales tanto en ese primer control como en los sucesivos, por lo que no se realizaron más pruebas complementarias, salvo seguimiento ecográfico estrecho.
En el ecocardiograma posnatal, la mayoría de casos de VCSIP son diagnosticado a través de un seno coronario dilatado visualizado en los planos de 4 cámaras y eje para esternal largo (confirmándose con el test de burbujas); y la ausencia de VCSD al no visualizarse en el plano subcostal eje de cavas, la presencia de una vena cava superior, objetivándose solo la entrada de la VCI en la AD.
La confirmación ecocardiográfica posnatal de la VCSIP junto con ausencia de VCSD es de vital interés para descartar cardiopatías congénitas asociadas de difícil diagnóstico durante la época fetal, como el drenaje venoso anómalo o la coartación de aorta además de su asociación con cromosomopatías o síndromes.
Previa a esta confirmación ecocardiográfica que será llevada a cabo por el cardiológico infantil, cabe destacar la actuación del pediatra general en la primera exploración física y en la anamnesis a la familia para el conocimiento de antecedentes familiares (tales como cardiopatías congénitas, síndromes familiares, cromosomopatías, etcétera) y personales de la madre (consumo de tóxicos o medicamentos durante el embarazo, diabetes gestacional, infecciones connatales, etcétera).
Durante la exploración por aparatos en las primeras horas de vida, será importante excluir rasgos dismórficos o alteraciones en la inspección y auscultación cardiopulmonar que nos puedan sugerir o hacer sospechar la presencia de posibles cardiopatías congénitas críticas o síndromes asociados.
Por tanto, el diagnóstico prenatal de la VCSIP junto con la agenesia de VCSD puede permitir tener un alto grado de sospecha al pediatra durante la primera exploración física del recién nacido, de posibles alteraciones asociadas y entonces poder llevar a cabo una actuación clínica precoz.
De igual manera, a pesar de que esta anomalía del sistema venoso sistémico se describa como una alteración anatómica benigna, puede tener importantes implicaciones clínicas posteriores tales como, dificultad a la hora de contar con accesos vasculares permeables para la canalización de vías venosas centrales o colocación de marcapasos o desfibriladores (pudiendo insertarse en zonas anómalas ante el desconocimiento de su existencia en la arteria subclavia, arteria carótida, mediastino, pericardio o espacio pleural).
Otra posible implicación es que en los casos que asocien defectos del septo interauricular puede existir un riesgo incrementado de embolias sistémicas paradójicas y por tanto de accidentes cerebrovasculares debido a la presencia de un shunt de derecha a izquierda2,8.
En nuestros dos casos, tras una correcta anamnesis donde se descartaron antecedentes familiares o personales maternos de relevancia y tras una primera exploración física normal en las primeras 24 horas de vida llevada a cabo por el neonatólogo encargado de la planta de maternidad, se confirmó ecocardiográficamente, mediante interconsulta al cardiólogo infantil la ausencia en ese momento de otras cardiopatías congénitas/estructurales asociadas.
Bien es verdad que en el primer caso se observó en el ecocardiograma posnatal una CIA OS pequeña, que, en los controles sucesivos durante el primer año de vida confirmaron que se trataba de un foramen oval permeable. De igual manera, la válvula mitral presentó un desarrollo normal sin insuficiencias y con un flujo transmitral normal en todo momento a pesar de una primera descripción como válvula displásica. Los ECG posteriores de control tampoco presentaron alteraciones en ningún momento.
En el segundo caso, a pesar de no presentar cardiopatía estructural ni alteraciones electrocardiográficas en el primer control posnatal, se continuó realizando seguimiento los 12 meses sucesivos, confirmando la ausencia de patología asociada.
Conclusiones
Si durante las ecografías morfológicas fetales se diagnostica esta alteración combinada del sistema venoso sistémico, es preciso realizar una ecografía morfológica detallada junto con ecocardiografía fetal dada la asociación con anomalías cardiacas y extracardiacas.
Ante su hallazgo aislado, a pesar de ser una anomalía venosa con un pronóstico perinatal favorable, cabe destacar la importancia de su confirmación posnatal para descartar cardiopatías de difícil diagnóstico durante la época fetal tales como el drenaje anómalo venoso parcial o la coartación de aorta.
De igual manera, su diagnóstico prenatal facilitará al pediatra tener un alto grado de sospecha de posibles síndromes o CC asociadas que permitirán realizar una actuación clínica precoz.
Así mismo, a pesar de tratarse de una patología venosa “benigna” variante de la normalidad, tiene importantes implicaciones clínicas tales como el posible desarrollo de taquiarritmias y posibles embolias sistémicas, dificultad en la canalización de vías centrales, colocación de dispositivos tales como desfibriladores o marcapasos.

