











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
La colestasis se define como la reducción en la formación o el flujo de bilis que da como resultado la retención de sustancias biliares dentro del hígado que normalmente se excretan en la bilis y se eliminan en la luz intestinal1.
Las recomendaciones internacionales sugieren realizar medición de bilirrubina sérica total y directa para detectar colestasis a todo lactante alimentado con fórmula o preparados que presente ictericia después de las 2 semanas de edad, o 3 semanas en pacientes alimentados con lactancia materna exclusiva y que presentan buen estado de salud. La hiperbilirrubinemia directa >1,0 mg/dl (17 mmol/l) se considera patológica y justifica continuar la evaluación diagnóstica1.
Son múltiples las causas de colestasis en pediatría, entre las que se incluyen anomalías estructurales que causan obstrucción al flujo biliar intra o extrahepático; causas infecciosas; tóxicas o metabólicas que alteran los mecanismos de síntesis y excreción de las sales biliares. Las etiologías más frecuentes de colestasis neonatal son: atresia de vía biliar, hepatitis neonatal idiopática, infecciosas, secundarias a nutrición parenteral, déficit de 1-antitripsina, metabólicas (tirosinemia y galactosemia), síndrome de Alagille y colestasis intrahepática familiar progresiva2-4.
La valoración inicial de los pacientes con colestasis neonatal debe ir dirigida al diagnóstico temprano de patologías tratables. Por sus implicancias pronósticas el pediatra debe, en primera instancia, descartar la atresia de la vía biliar, dado que su tratamiento médico-quirúrgico precoz se asocia con mejor pronóstico. Otras patologías como sepsis, hipotiroidismo, panhipopituitarismo y errores innatos del metabolismo como la galactosemia y la tirosinemia también deben considerarse por requerir tratamientos específicos1-4.
El síndrome de Alagille, es una patología poco frecuente que afecta a uno de cada 100.000 recién nacidos vivos5. Se trata de una ductopenia sindrómica caracterizada por colestasis crónica, progresiva. Es una enfermedad genética, de transmisión autosómica dominante, con expresión variable, secundaria generalmente a mutaciones en el gen JAG1. Un pequeño número de pacientes presentan mutaciones en el gen NOTCH26. Como consecuencia, se inducen alteraciones del desarrollo embriológico que afectana estructuras dependientes del mesodermo, como el aparato cardiovascular, hígado y vasos sanguíneos. Entre las mutaciones encontradas, 50%-70% son esporádicas o de novo, mientras que 30%-50% son hereditarias7.
Los primeros casos de síndrome de Alagille fueron descritos en 1973 por Watson y Millar, identificando la relación existente entre enfermedad hepática neonatal y estenosis pulmonar. El síndrome de Alagille se expresa con afectación multisistémica y variabilidad fenotípica, existen formas subclínicas y presentaciones con manifestaciones cardíacas, hepáticas o renales graves7,8. Las manifestaciones clínicas más frecuentes son la colestasis asociada a prurito y xantomas, el compromiso cardíaco con estenosis pulmonar (pueden existir otras anomalías como tetralogía de Fallot, defectos septales, entre otras), alteraciones vertebrales (vértebras en alas de mariposa), oculares (embriotoxon posterior), hipoacusia, dismorfias faciales características (facies triangular, puente nasal ancho, ojos hundidos), renal (displasia renal) y talla baja. Asimismo, pueden asociar malformaciones vasculares sistémicas o cerebrales. Los estudios de correlación genotipo-fenotipo, no han mostrado asociación entre el tipo de mutación y la gravedad de la expresión clínica9.
Las manifestaciones clínicas aparecen entre los 2 y 3 meses de edad, relacionadas generalmente con la colestasis, realizándose el diagnóstico en los primeros meses de vida en la mayoría de los casos. El diagnóstico es clínico, pudiendo confirmarse mediante estudios genéticos si estos se encuentran disponibles; la biopsia hepática si bien no es imprescindible se mantiene como un recurso de apoyo a la sospecha clínica10,11.
El abordaje terapéutico suele ser interdisciplinario e individualizado. El mismo estará enfocado en el control sintomático, la prevención de complicaciones de la colestasis (malnutrición y deficiencia de vitaminas liposolubles), así como tratar las malformaciones asociadas.
El objetivo de esta comunicación es describir el abordaje diagnóstico y terapéutico de un lactante pequeño con colestasis secundaria al síndrome de Alagille.
Caso clínico
Lactante de 2 meses, sexo femenino. Producto de embarazo mal controlado en cantidad y calidad. Sin antecedentes perinatales patológicos. No presentó ictericia en período neonatal. Buen crecimiento y desarrollo. Bien controlado en salud. Sin antecedentes familiares a destacar.
Consulta por ictericia de 10 días de evolución, progresiva, acompañada de hipocolia y coluria. En apirexia, sin prurito. No ingesta de tóxicos ni tisanas.
Al examen físico se encontraba reactiva, dismorfias faciales dadas por cara triangular, hipertelorismo, puente nasal ancho y frente prominente. Bien nutrida. Ictericia hasta tobillos y compromiso de palmas. Hemodinamia estable, soplo sistólico 3/6 en todo el precordio, pulsos y presión arterial normales. Abdomen sin visceromegalias. Deposiciones hipocólicas en pañal (3-4 según cartilla colorimétrica de heces) y coluria.
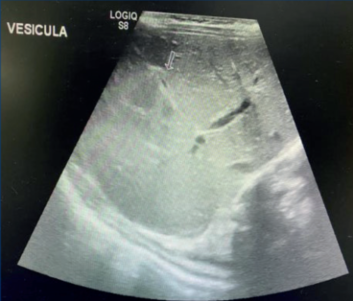
Con planteo clínico de colestasis se solicitaron estudios de laboratorio e imagen para valorar afectación multisistémica y pesquisa etiológica. Los estudios complementarios solicitados evidenciaron presencia de hiperbilirrubinemia (14,9 mg/dl) a predominio de la bilirrubina directa (13,7 g/dL), fosfatasa alcalina (FA) aumentada (698 U/L), Gamma glutamil transferasa (GGT) (508 U/L) e hipercolesterolemia (327 mg/dL) lo cual traduce un patrón colestásico. Como parámetro de lesión hepática, las transaminasas, aspartato aminotransferasa (TGO) y alanina aminotransferasa (TGP), presentaban un aumento moderado (242 U/L, 203 U/L) respectivamente. Los estudios sobre la función hepática fueron normales (crasis sanguínea, glicemia, albuminemia, amoniemia). La ecografía abdominal destacaba la presencia de una vesícula biliar contraída, atrófica, hígado y bazo sin alteraciones con vía biliar principal intra y extrahepática no dilatada (Figura 1).
Para el estudio etiológico de la colestasis se solicitaron serologías para virus de hepatitis A, B, C, virus de la inmunodeficiencia humana (HIV), VDRL, virus herpes simple 1 y 2, anticuerpos (Ac) anti toxoplasmosis IgM e IgG, Ac anti citomegalovirus (CMV) IgM e IgG, Ac anti rubéola IgM que fueron no reactivos. La dosificación de alfa 1 antitripsina fue normal. La pesquisa neonatal con tripsina inmunorreactiva, test de sudor, glucemia y función tiroidea fueron normales.
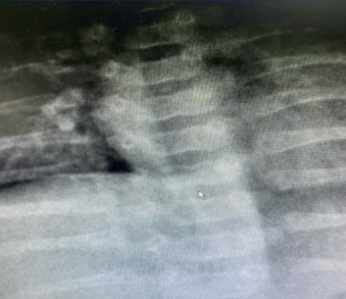
Se indicaron estudios para evaluar afectación multisistémica: radiografía de columna vertebral: cuerpos vertebrales dorsales D7 y D11 con morfología sugestiva de vértebras “en mariposa” (Figura 2). Ecocardiograma: estenosis leve de rama pulmonar derecha. Valoración oftalmológica: embriotoxon posterior en ambos ojos, fondo de ojo sin alteraciones. Biopsia hepática: colestasis neonatal de patrón mixto, ductopenia, índice ducto/espacio porta 2/28 y hepatitis neonatal.
Se inició tratamiento con ácido ursodesoxicólico y vitaminas liposolubles (A, D, E y K).
Presentó buena evolución clínica, con reducción de ictericia y descenso de bilirrubina directa, otorgándole alta a los 17 días de hospitalización con seguimiento ambulatorio por pediatra y gastroenterólogo pediatra.
Discusión
La paciente del caso clínico reportado presentaba colestasis clínica dada por ictericia, hipocolia y coluria. Las enfermedades que se presentan con colestasis en el lactante son múltiples8. En este caso, se solicitaron en primera instancia estudios de laboratorio para confirmar la colestasis, encontrando hiperbilirrubinemia a predominio de la bilirrubina directa con aumento de la FA y GGT e hipercolesterolemia, un patrón colestásico. Simultáneamente se avanzó en el estudio etiológico. El primer estudio imagenológico a solicitar es la ecografía abdominal. Varios parámetros ecográficos hepáticos, como el signo del cordón triangular, la morfología vesicular anormal, falta de contracción de la vesícula biliar después de la alimentación oral, falta de visualización del conducto biliar común, diámetro de la arteria hepática, relación entre el diámetro de la arteria hepática y el de la vena porta y el flujo sanguíneo subcapsular se ha sugerido como elementos orientadores de atresia biliar. Es imperativo recordar que una ecografía abdominal normal, sin embargo, no descarta la atresia biliar1,6,8,11.
El estudio ecográfico evidenció una vesícula presente, que, en el contexto de este paciente, con características de síndrome de Alagille, aleja el planteo de atresia de vía biliar.
Se descartaron también causas intrahepáticas de colestasis como la etiología infecciosa y el déficit de alfa 1 antitripsina. Las causas endocrinológicas y metabólicas más comunes (por no tener fallo hepático y tener glucemias normales) se excluyeron. Teniendo en cuenta que la paciente presentaba, además, dismorfias faciales y estenosis leve de la rama pulmonar derecha, se dirigieron los estudios a confirmar el probable síndrome de Alagille.
El adecuado y oportuno diagnóstico de este síndrome tiene implicancias pronósticas. La literatura refiere que los casos que generaron dudas diagnósticas respecto al diagnóstico diferencial con atresia biliar y fueron sometidos a cirugía de Kasai el pronóstico fue peor12.
El diagnóstico de síndrome de Alagille se establece con los criterios diagnósticos clínicos y/o mediante estudios genéticos moleculares. La colestasis crónica asociada a ductopenia o ausencia de ductus biliares intrahepáticos, facies peculiar, anomalías vertebrales, anomalías oftalmológicas (principalmente embriotoxon posterior) y cardiopatía congénita (especialmente estenosis pulmonar periférica) son los cinco criterios diagnósticos mayores, los cuales estaban presentes en la paciente5,8.
La literatura describe como las dismorfias faciales más reportadas en este síndrome al hipertelorismo, cara triangular, frente amplia y prominente, ojos profundos, nariz recta, puente nasal amplio, aplanamiento malar, maxilar inferior pequeño y mentón puntiagudo, muchas de las cuales fueron encontradas en este caso5,7,8,13.
En cuanto a las características cardíacas, más del 90% de los casos tienen anomalías, siendo la estenosis de las ramas pulmonares la más frecuente. Otras lesiones cardíacas encontradas son la tetralogía de Fallot y los defectos septales, en menor proporción14,15.
Los hallazgos en el sistema esquelético más reportados son las vértebras torácicas en forma de alas de mariposa. Se deben a fusiones anormales de la columna vertebral que conducen a hendiduras sagitales en 80% de los casos, tal como se evidencia en esta paciente.
Dentro de los hallazgos oftalmológicos, el más frecuente es el embriotoxón posterior (79%-89%), sin afectación de la agudeza visual. Otros defectos oculares que pueden llegar a presentarse incluyen la anomalía de Axenfeld-Rieger, retinosis pigmentaria, y anomalías papilares y del disco óptico8,13,15,16.
Pueden existir otras anomalías, consideradas como menores: alteraciones renales estructurales y/o funcionales (40%), anomalías vasculares, xantomas, afectación de peso y talla, infecciones pulmonares recurrentes, insuficiencia pancreática, hipotiroidismo, anomalías vasculares, alteraciones neurológicas, hipogonadismo y pubertad retrasada, retraso mental, afectación del tono vocal5-7,13,14,17,18.
El diagnóstico del síndrome de Alagille se puede confirmar mediante biopsia hepática, la cual muestra habitualmente colestasis crónica y escasez de conductos biliares intrahepáticos. Se puede evidenciar también fibrosis de grado variable. Estos hallazgos suelen ser progresivos con la edad, en lactantes menores de seis meses pueden no estar presentes7,13.
El pilar genético consta de la realización de pruebas moleculares, detectando mutaciones en los genes JAG1 o NOTCH2, disponibles en Uruguay5,7,18.
El estudio realizado por Gilbert y colaboradores en 2019 sobre las mutaciones genéticas en el síndrome de Alagille, encontró que 94,3% de las personas con este síndrome clínico tenían una variante patológica en el gen JAG1, 2,5% en el gen NOTCH2 y en 3,2% no se encontraron mutaciones19.
Por otra parte, existe la posibilidad de realizar diagnóstico prenatal con pruebas genéticas moleculares, siendo también útil el ultrasonido fetal, especialmente el ecocardiograma fetal, para detectar un defecto estructural cardíaco significativo17. En el caso presentado el resultado de biopsia hepática que evidenció escasez de conductos biliares intrahepáticos apoyó el planteo diagnóstico. Teniendo los criterios clínicos y la biopsia hepática compatible, no fue necesaria la secuenciación de los genes para confirmar el síndrome de Alagille.
El abordaje terapéutico requiere un enfoque multidisciplinario, dependiendo de los hallazgos identificados. No existe un tratamiento específico, se basa en el control sintomático, especialmente del prurito, realizar un adecuado soporte nutricional y la suplementación con vitaminas liposolubles (A, D, E, K)20-22.
Para tratar el prurito severo y los xantomas se utilizan agentes que favorecen el flujo biliar (ácido ursodesoxicólico, naltrexona, rifampicina y colestiramina). En los últimos años se han incorporado nuevas estratégias farmacológicas, cuya evidencia se viene consolidando23-25.
En un ensayo aleatorizado en 29 niños con síndrome de Alagille, maralixibat, un inhibidor del transporte intestinal de ácidos biliares, redujo los ácidos biliares séricos medios y mejoró los síntomas de prurito, la calidad de vida, el crecimiento lineal y los xantomas, y fue bien tolerado. Estos hallazgos motivaron la aprobación de maralixibat por parte de la Administración de Drogas y Alimentos de los EE.UU. para el tratamiento del prurito colestásico en el síndrome de Alagille24,25.
La derivación biliar interna parcial quirúrgica y la exclusión ileal también se han utilizado para este propósito, sin prevenir la progresión de la enfermedad hepática16,17. En este caso se administraron suplementos vitamínicos liposolubles y ácido ursodesoxicólico.
La afectación cardíaca, renal y vascular se maneja de acuerdo con los síntomas y alteraciones existentes. Las anomalías oftalmológicas y vertebrales no suelen necesitar intervención9-11,13,15-17.
En el futuro, los nuevos enfoques terapéuticos pueden implicar la modulación de la señalización de la vía Notch, terapias celulares o la corrección de mutaciones específicas in vitro o in vivo26.
El pronóstico de esta enfermedad a largo plazo depende de la severidad del daño hepático y las malformaciones cardiovasculares asociadas. Entre 21% y 31% de los pacientes requieren trasplante hepático por el desarrollo de cirrosis con insuficiencia hepática, hipertensión portal o prurito incoercible. Un 15% puede desarrollar complicaciones como hepatocarcinoma, insuficiencia hepática, insuficiencia pancreática exocrina o fibrosis hepática con hipertensión portal. La mortalidad es aproximadamente de 10%, debido a fallo cardíaco o hepático severo. Es importante a largo plazo la monitorización de la función cardíaca y renal, así como el cribado para el desarrollo de carcinoma hepatocelular7,10,13,15,16,18,20,26.
Conclusiones
En todo lactante pequeño que presenta colestasis es necesaria una evaluación minuciosa con el objetivo de determinar su etiología. Algunas de sus causas pueden poner en riesgo la vida y requerir un abordaje médico-quirúrgico oportuno. En este caso se visualizaron todos los criterios diagnósticos mayores del síndrome de Alagille, confirmándose esta entidad con el estudio anatomopatológico. Es prioritario el abordaje y seguimiento interdisciplinario.

