











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Introducción
El síndrome “incisivo central maxilar medio único, baja estatura, síndrome de atresia de coanas/ estenosis medio-nasal”, ahora abreviado como “síndrome del incisivo central maxilar medio único” (SMMCI), debido a que todas estas características no tienen que darse necesariamente en todos los casos, fue descrito por primera vez por Scott en 19581.
Es una entidad rara (con una incidencia de 1:50.000 nacidos vivos) que es reconocida por la presencia de un hallazgo dental aislado (erupción de un único incisivo central como dato patognomónico) o como parte del llamado síndrome SMMCI, que se produce por factores desconocidos que actúan en el útero entre los días 35 y 38 a partir de la concepción, involucrando huesos craneales, maxilar, vías respiratorias (atresia coanal, estenosis nasal media y estenosis de la apertura piriforme nasal (considerada diagnóstica como una medición inferior a 11 mm a nivel del meato inferior en un lactante a término)) y estructuras de la línea media cerebrales1,2.
El diagnóstico se realiza habitualmente postnatalmente, sobre los 8 meses de edad con la erupción de un único incisivo central. Siendo un diagnóstico prenatal y en periodo neonatal posible, aunque más difícil.
Presentamos el caso de un neonato diagnosticado de SMMCI, que debutó con dificultad respiratoria y problemas en la alimentación tras el nacimiento.
Caso clínico
Mujer de 21 horas de vida que ingresa en Unidad de Neonatología por saturación de oxígeno del 89% coincidiendo con la toma. A la exploración física destaca una columela con punta nasal lateralizada hacia la izquierda, cornaje nasal y tiraje subcostal intermitente secundario a moldeamiento nasal. Tonos rítmicos sin soplos, con buena entrada de aire bilateral. Saturación de oxigeno del 100% en el momento de nuestra valoración, con adecuada coloración cutáneo-mucosa. Se introduce por narinas con mínima resistencia una sonda del número 5 para descartar atresia de coanas. El neonato es valorado por otorrinolaringología, quienes tras comprobar lateralización de la columela sin repercusión en tamaño de vestíbulo nasal, aconsejan actitud expectante ante la realización de fibroscopia.
Como antecedentes personales, nace con una edad gestacional de 39+0, con un peso de 2.420 g (p3), talla 46 cm (p3), perímetro cefálico 33 cm (p3-10). En las ecografías prenatales se detecta feto con crecimiento intrauterino restringido (CIR) tipo I por Doppler patológico de arterias uterinas. Nace por cesárea urgente por alteración del registro cardiotocográfico. Apgar 8/10/10.
Madre y padre de 37 y 39 años respectivamente, sanos. Niegan enfermedades hereditarias conocidas en la familia.
El neonato es dado de alta tras 48 horas en la unidad tras comprobar adecuadas tomas de lactancia materna, desaparición del distrés respiratorio con persistencia del cornaje nasal secundario a probable moldeamiento. Es citado en consultas de pediatría para seguimiento.
A los 16 días de vida es remitido desde consulta de pediatría por una pérdida de peso respecto al nacimiento del 15% (2.050 g), por lo que ingresa por desnutrición secundaria a succión no efectiva y deshidratación isonatrémica. Es dada de alta tras recuperación del peso a los 22 días de vida bajo control estrecho por pediatra.
A los 50 días de vida consulta en urgencias por dificultad respiratoria y rechazo de las tomas secundarios a infección respiratoria superior agravada por la anatomía de la paciente. A la exploración física se evidencia paladar ojival en V, hipotelorismo, raíz nasal plana con anteversión de narinas (ya objetivados en controles de seguimientos previos, más evidentes con el crecimiento de la paciente). Se decide ampliar estudio con TAC de cabeza y senos paranasales donde se objetiva estenosis de orificios piriformes (Figura 1) (derecho de 4 mm, izquierdo de 1,2 mm) por medialización del proceso nasal del hueso maxilar que asocia desviación del tabique nasal al lado izquierdo junto a paladar duro hipoplásico con morfología triangular y megaincisivo central único en maxilar superior (Figura 2). Se realiza resonancia magnética (RM) cerebral donde no se visualizan otras alteraciones en cerebro, cerebelo o tronco con hipófisis presente en silla turca de aspecto normal. Se realiza nasofibrolaringoscopía objetivando estrechez de narinas y fosas nasales que dificulta el paso del fibroscopio hasta laringe. Radiografía de tórax y serie ósea, ecografía abdominal, ecocardiografía sin alteraciones. Estudio analítico (incluido perfil tiroideo) normal. Es dada de alta con alimentación por sonda nasogástrica bajo seguimiento estrecho en consultas de dismorfología, donde se completa estudio genético con cariotipo 46 XX y CGH Array en el que no se detectan variaciones de cambio de número de copia de naturaleza no polimórfica.
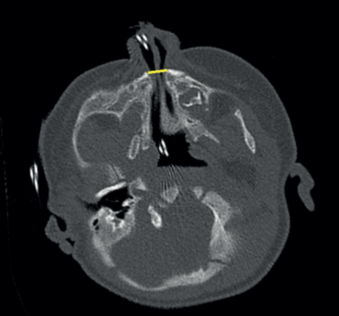
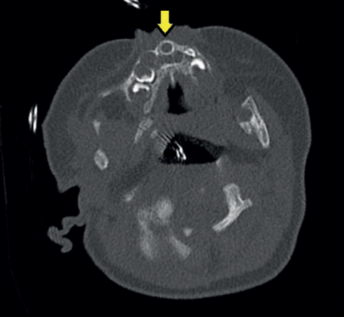
La paciente es derivada para valoración y seguimiento por neurología y endocrinología pediátricas, así como cirugía maxilofacial, quienes intervienen a la paciente realizando permeabilización de orificios piriformes a los 5 meses de edad. Actualmente la paciente mantiene una buena evolución clínica, con adecuada ganancia pondoestatural para la edad.
Discusión
La etiología del SMMCI es incierta. Las causas más comunes son mutaciones heterocigotas en el SHH (Sonic Hedgehog) en el cromosoma 7q36. SHH codifica una proteína que es fundamental en el modelado del embrión. Hay un total de 36 variantes genéticas con 7 genes que están relacionados, publicados. La mayoría de variantes se encuentran en el gen SHH (77,7%). También se encontraron mutaciones en los genes: SIX3 (8,3%), TGIF1 (2,7%), GLI2 (2,7%), PTCH1 (2,7%), SALL4 (2%) y FGF8 (2,7%). Los casos de SMMCI causados por anomalías cromosómicas se han asociado hasta ahora con los cromosomas: 7 (del (7q36 ter)), 18 (del (18p) yr (18)), 22 (del (22q11.2)) y 47 (47XXX). Las deleciones 18p y 7q son las más frecuentes3.
El SMMCI puede ser fenotípicamente muy variado, lo que puede dificultar su diagnóstico. En los neonatos, debe sospecharse en el caso de niños con coanas estrechas y obstrucción nasal (presente en el 90% de los casos al nacimiento), como exponemos con nuestro caso, en especial en aquellos con mandíbula superior prominente, paladar arqueado y/o ausencia de frenillo lingual. Si hay algún grado de hipotelorismo, debe considerarse su asociación con holoprosencefalia y sus diferentes variantes, quedando descartada mediante RM cerebral en nuestra paciente.
Pueden asociarse otras anomalías congénitas, como cardiopatías (25%), labio leporino y/o paladar hendido, microcefalia (33%), hipopituitarismo e hipotiroidismo (25%), hipotelorismo (45%), estrabismo convergente, atresia esofágica y duodenal (10%), hemivértebras cervicales y/o escoliosis (14%), hipoplasia de clavículas, agenesia renal, quiste dermoide cervical, micropene y genitales ambiguos. Más frecuente en parto prematuro y bajo peso al nacer (en 37%) como fue nuestro caso y en 14% de madres con diabetes gestacional. Durante su evolución podemos detectar baja estatura en 50% de los niños (requiriendo hormona de crecimiento hasta 33%), así como discapacidad intelectual (50%)3,4.
Así pues, puede ocurrir como una anomalía aislada o en asociación con otros síndromes como son: CHARGE, VACTERL, velocardiofacial, holoprosencefalia (autosómico dominante), displasia de ectodermo y síndrome de retracción de Duane3,4.
El diagnóstico es posible con ultrasonido entre las 18-22 semanas (siendo esencial un estudio prenatal minucioso del maxilar superior, en especial en aquellos fetos con malformaciones cerebrales o faciales) o, posiblemente, en pruebas genéticas en casos familiares, pero rara vez se hace antes del nacimiento por pasar desapercibido en el estudio ecográfico, como fue nuestro caso. Con el conocimiento actual de la condición, el diagnóstico debe ser hecho a más tardar en el momento de la erupción del diente incisivo maxilar primario que se produce aproximadamente a los 8 meses de edad2,4.
El tratamiento será sintomático e individualizado según las características de nuestro niño. El tratamiento de la estenosis nasal del piriforme debe basarse en la gravedad de los síntomas. En casos con dificultad respiratoria persistente, si el tratamiento conservador resulta ineficaz, con pobre ganancia de peso y en niños con un ancho de apertura igual o menor de 5,7 mm, se debe considerar un tratamiento quirúrgico para ensanchar la entrada ósea. Aunque en niños muy pequeños, como los neonatos, debido a la plasticidad natural de las estructuras óseas y cartilaginosas del esqueleto maxilofacial se puede considerar un enfoque más conservador como el realizado. Por su parte, el incisivo central solo produce un problema estético, que se trata de manera ideal mediante ortodoncia, prostodoncia y/o cirugía; aunque de forma alternativa, puede dejarse sin tratar, dependiendo de las preferencias del paciente o su familia. La baja estatura puede necesitar terapia con hormona de crecimiento u otra terapia hormonal sustitutiva dependiendo del déficit hormonal según los casos de hipopituitarismo5,6.
Por lo tanto, un abordaje multidisciplinar por pediatras, neurólogos, cardiólogos, endocrinólogos, genetistas, otorrinolaringólogos, cirujanos maxilofaciales y odontólogos es necesario para el adecuado manejo de estos niños. El asesoramiento genético tanto del paciente como de su familia es importantes para confirmar el diagnóstico como para planificar futuras gestaciones7.
Conclusiones
El SMMCI puede presentarse como un hallazgo clínico único o como parte de un síndrome que puede afectar a otras estructuras de la línea media y a multitud de órganos siendo característico su asociación con talla baja, obstrucción nasal congénita y holoprosencefalia que debe sospecharse especialmente en niños con hipotelorismo. Puede ser diagnosticado prenatalmente mediante ecografía, neonatalmente debiéndolo descartar siempre en neonatos con obstrucción nasal congénita como en el caso expuesto, así como en la infancia, nunca después de los 8 meses de vida, con la erupción del incisivo central maxilar único, dato patognomónico de esta entidad. El manejo será multidisciplinar por parte de pediatras, otorrinolaringólogos, genetistas y dentistas, entre otros.

