











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Introducción
Los niños con síndrome de Down (SD) presentan características fenotípicas específicas junto con diversas malformaciones asociadas, como cardíacas, digestivas, cerebrales, renales y oculares, entre otras. Un 60% de las malformaciones en el SD suelen ser las oculares. Entre ellas, las ametropías (miopías y astigmatismo), queratocono y las cataratas son las de mayor prevalencia. A su vez, estas últimas son clasificadas según su tiempo de aparición en congénitas, de la infancia, la adolescencia y la adultez, con una variabilidad de presentación entre 3% a 20% tanto unilaterales como bilaterales. El porcentaje de cataratas congénitas (CC), representa de 3% a 5% siendo así la presentación menos frecuente dentro de este síndrome1.
Según su constitución morfológica las CC se clasifican en: polar anterior, polar posterior, centrales (pulverulento, nuclear total, zonular o lamelar) y totales. Cuanto más central y posterior es la CC respecto a la mácula más ambliopizante es, es decir presenta mayor compromiso visual, por lo tanto requiere una resolución quirúrgica más rápida2. Cabe destacar también que el mayor compromiso del eje visual y la extensión de la catarata implica priorizar la resolución quirúrgica temprana. Según su extensión topográfica, las CC de ubicación central representan más transcendencia que las de ubicación periférica. La Sociedad Argentina de Oftalmología Infantil las clasifica según su diámetro menor o mayor a 2,5 mm, siendo estas últimas todas indicaciones quirúrgicas tempranas.
La CC bilateral es una prioridad oftalmológica que debe ser operada tempranamente en los primeros meses de vida, ya que con posterioridad la ambliopía por deprivación generará nistagmus, pronóstico visual reservado y baja agudeza visual final. Su identificación temprana, diagnóstico y adecuado tratamiento es la clave para obtener resultados óptimos3. Representa un desafío para el médico especialista, no solo por su resolución quirúrgica a corto plazo sino por su seguimiento y manejo posoperatorio, que es de suma relevancia para su tratamiento.
Caso clínico
Paciente de sexo masculino, edad gestacional 38 semanas, con peso adecuado, madre con 41 años, tercigesta, con dos hermanas por vía materna sanas, embarazo controlado, sin antecedentes prenatales de relevancia presentando ecografía de las 12 semanas con translucencia nucal en rango normal y serologías para HIV, HBV, toxoplasmosis, sífilis y Chagas no reactivas. Nace por cesárea en otra institución. Diagnóstico clínico post natal de SD. Se realiza fondo de ojo donde se observa opacidad cristalineana con reflejo rojo (RR) negativo con diagnóstico clínico de CC bilateral. No presenta antecedentes familiares de cataratas. Fue otorgada el alta sanatorial a los tres días de vida, sin seguimiento adecuado.
Ingresa a nuestra institución al quinto día de vida por hiperbilirrubinemia y regular progreso de peso. Durante la internación se inicia el tratamiento del paciente, su evaluación y enfoque multidisciplinario, incluyendo el servicio de oftalmología, genética, cardiología, diagnóstico por imágenes, infectología y endocrinología. Se solicitan serologías para herpes S., sarampión, parvovirus 19 y rubéola, que resultan no reactivas, y citomegalovirus en orina con resultado negativo. Es evaluado por genética clínica, que informa fenotipo compatible con SD, cariotipo bandeo G, 47, XY, +21. A nivel oftalmológico se procede a una examinación externa con lupa de 28 dioptrías, biomicroscopia, fondo de ojo y se realiza como método complementario ecografía para evaluación del segmento posterior del ojo, concluyendo en el diagnóstico de catarata nuclear central bilateral (Figura 1) y (Figura 2). A nivel sistémico no se detecta otro tipo de malformaciones o alteraciones asociadas al SD. Se otorga egreso sanatorial al octavo día de vida con seguimiento oftalmología infantil. Se realiza cirugía de cataratas congénitas a las 9 semanas y 12 semanas de vida, ojo derecho e izquierdo respectivamente, procedimiento bien tolerado, sin complicaciones. La intervención realizada fue lensectomía, capsulotomía posterior y vitrectomía anterior en ambos ojos. Posterior a la intervención se indicó lentes de contacto, rehabilitación visual y seguimiento programado. La colocación de un lente intraocular, debido al crecimiento ocular continuo y sus complicaciones, no es recomendado antes del año de vida, se programará con posterioridad.
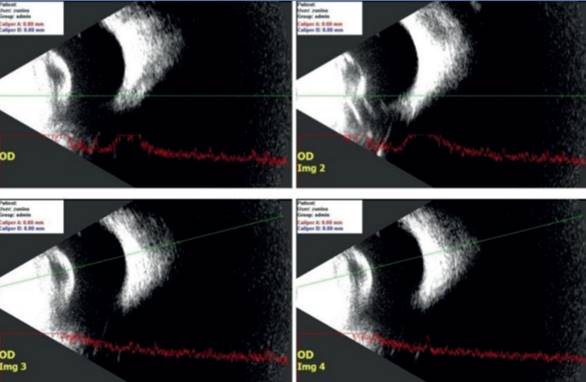
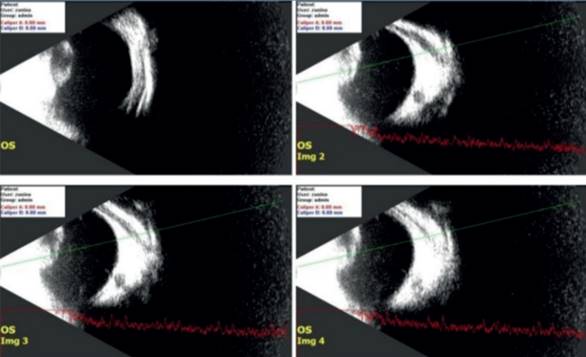
Figura 1: Cavidad vítrea acústicamente vacía. Se detecta marcado aumento de reflectividad en la cápsula posterior del cristalino. No se detecta banda de fibrosis retrolental, solo espesamiento capsular. Cuadro ecográfico compatible con catarata congénita
Discusión
La prevalencia global de CC se encuentra estimada de 1 a 4 por cada 10.000 niños en países desarrollados y de 5 a 15 por cada 10.000 niños en países en vías de desarrollo. Representa 13% de las causas de disminución visual en niños y se considera como la causa más frecuente de deprivación visual tratable. En América Latina se estima que 1 de cada 300 nacimientos por año presenta CC y es responsable del 20% de las causas de ceguera en la infancia4.
Las CC resultan de múltiples causas, tales como historias familiares de CC, enfermedades infecciosas asociadas tipo TORCHS, enfermedades metabólicas y síndromes genéticos, sobre todo el SD3.
La asociación de cataratas con SD es variable, presentándose en la infancia, adolescencia y en la adultez, siendo la CC bilateral de aparición temprana la menos frecuente.
La visualización del perfil facial, las órbitas y los cristalinos forman parte de la recomendación para la evaluación de la cara fetal en el examen rutinario de la ecografía morfológica del segundo trimestre o scan fetal realizado durante el embarazo. Se resalta la facilidad de la técnica y su observación prenatal, que conlleva a una pérdida del diagnóstico oportuno5.
En el recién nacido se presenta como leucocoria (pupila blanca), que es la alteración o ausencia del RR. En condiciones normales este reflejo (test de Bruckner) se obtiene en un ambiente semioscuro con un oftalmoscopio a una distancia de 30 cm, es rojizo y simétrico. Debido a la facilidad de este testeo y su sencillez, puede ser realizado en todo recién nacido por un pediatra o médico de atención primaria. Se recomienda efectuar la exploración RR antes del alta de neonatología, y de presentar un RR anormal o ausente su derivación temprana a un oftalmólogo6,7.
Estos procedimientos resultan imprescindibles para el diagnóstico precoz ya que son sencillos, y el no realizarlo conlleva a consecuencias irreversibles como la ambliopía.
Al presentar un RR ausente, con fondo de ojo sin visualización debido a la catarata nuclear central bilateral, la ecografía es una herramienta muy útil para la valoración clínica y el diagnóstico de procesos que afectan el polo posterior del ojo. Permite también descartar asociaciones con otras patologías8.
Una vez diagnosticado, es crucial la edad del paciente para realizar la cirugía, ya que muy tempranamente pueden presentar como complicación principal glaucoma, y después de una postergación nistagmus, estrabismo y llevar a la ambliopía9. Esta ambliopía, al ser causada por deprivación, justifica el inicio de las acciones tempranamente
Se recomienda realizar el procedimiento quirúrgico de las CC unilaterales entre las 6 y 8 semanas y las bilaterales entre las 6 y 10 semanas de vida respectivamente, con un periodo entre 1 a 3 semanas entre lensectomía para cada ojo9.
Los oftalmólogos se enfrentan a muchos desafíos en el manejo de CC, y los resultados no son a menudo los ideales Es importante comprender que la extracción de cataratas en un bebé no es un evento estático, pero tiene un impacto significativo sobre las estructuras oculares y su posterior crecimiento10,11. Resaltamos que el seguimiento posoperatorio es tan o más importante que la propia cirugía. Implica una gran complejidad debido al cambio de corrección óptica que presentan a lo largo del tiempo, el uso de gafas, lentes de contacto y su manejo tanto para el paciente como para los padres, y más en paciente con SD.
La CC precisa un enfoque multidisciplinario. Es importante el diagnóstico prenatal con ecografía, más en pacientes con riesgos asociados como cromosomopatías, enfermedades infecciosas tipo TORCHS, gestantes con historia familiar de cataratas congénitas u otras anomalías oculares. Realizar evaluación en la internación conjunta mediante un RR y evaluación posterior con oftalmología. El manejo de CC plantea desafíos únicos debido al desarrollo visual y al crecimiento ocular. La inmediata respuesta y el abordaje temprano permitirán darles mejores resultados a nuestros pacientes.

