











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
La disgenesia tubular renal es una entidad histológicamente caracterizada por ausencia o escaso desarrollo de los túbulos renales. Puede ser adquirida durante el período fetal o heredada con patrón autosómico recesivo. Clínicamente se manifiesta como oligoamnios, anuria fetal mantenida, alteraciones de la osificación craneal, insuficiencia renal neonatal, hipotensión e hipoplasia pulmonar. Presenta una alta mortalidad, dada principalmente por la anuria neonatal y la hipoplasia pulmonar.
La importancia del tema radica en tener conocimiento de esta patología, a pesar de ser poco frecuente, y considerarla como un posible diagnóstico diferencial.
La incidencia es difícil de estimar, ya que existen 70 casos reportados desde la primera descripción realizada en 1983 por Allarson y colaboradores1.
Caso clínico
Madre de 24 años, sin antecedentes patológicos a destacar. Tres gestaciones previas, dos partos vaginales, una cesárea por podálica. Tres recién nacidos vivos. Hija previa de 5 años de distinta pareja, con coartación de aorta, operada a los 12 días de vida.
Embarazo mal controlado, refiere cuatro controles, sin complicaciones.
Serologías para sífilis y VIH negativos. Exudado vaginorrectal para búsqueda de estreptococo del grupo B no realizado, ecografía estructural sin alteraciones.
Inicia trabajo de parto de 37 semanas, de manera espontánea, se decide operación cesárea por presentación podálica, rotura de membranas de tres horas de evolución, líquido amniótico claro, se envía placenta a anatomía patológica.
Se recibe recién nacido de 37 semanas de edad gestacional de sexo masculino, peso al nacer 2.560 g, longitud 48 cm, perímetro craneano 32 cm, Apgar 3/6/7. Gasometría de cordón sin acidosis.
Del examen físico en la recepción se destaca: depresión de la raíz nasal, cráneo con gran disyunción de suturas, impresiona multifracturado, hipospadias y criptorquidia bilateral, permeabilidad anal: sonda progresa hasta 4 cm.
Instala dificultad respiratoria a los 15 minutos de vida, ingresando a terapia intensiva. Asistencia ventiladora mecánica con altos requerimientos de oxígeno, requiere al tercer día de vida ventilación de alta frecuencia. Elementos sugestivos de hipoplasia pulmonar en la radiografía de tórax. Presentó múltiples complicaciones relacionadas con la ventilación: neumotórax, neumomediastino y sangrado pulmonar. Acidosis mixta persistente. Requerimiento de inotrópicos desde el nacimiento, dada hipotensión mantenida.
Se mantuvo en anuria desde el nacimiento con requerimiento de diuréticos. Falla renal, al tercer día de vida fue valorado por equipo de nefrología que plantea fuera de oportunidad para diálisis peritoneal.
Valorado por equipo quirúrgico se plantea atresia digestiva baja, rectal o colon descendente, fuera de oportunidad quirúrgica.
De la paraclínica realizada:
Ecocardiograma (del primer día de vida) que informa hipertensión pulmonar severa, ductus amplio con shunt de derecha a izquierda. Afinamiento de arco aórtico.
Ecografía renal y Doppler: riñones de tamaño normal, ambos con disminución moderada y difusa de la diferenciación corticomedular. Cavidades no dilatadas. Arterias renales permeables, Doppler normal, flujo intraparenquimatoso normal. Vejiga presente no distendida.
Ionograma sodio 144 meq/l, potasio 4,2 meq/l, calcio iónico 1,22 mmol/l, creatinina 3,26 mg/dl, azoemia 1,35 mg/dl.
Radiografía de abdomen e invertograma que informa distensión de asas intestinales, sin gases en porción distal. Acortamiento de huesos largos.
Ecografía abdominal: duodeno, asas y colon distendidos. Escaso líquido peritoneal.
Ecografía transfontanelar sin alteraciones.
Cariotipo: 46XY, patrón de bandeo.
Se discute con equipo de cuidados paliativos el primer día de vida, se decide limitación del esfuerzo terapéutico. Fallece al tercer día de vida.
Informe de la autopsia: dismorfias externas (raíz nasal deprimida), disgenesia tubular renal, hipospadias, hipoplasia pulmonar, falta de osificación de los huesos craneales hipocalvaria), asimetría de hemisferios cerebrales. Vinculados a hipoxia: hemorragia pulmonar, necrosis miocárdica focal, necrosis neuronal selectiva.
Discusión
La disgenesia tubular renal se caracteriza por la aparición, luego de las 22 semanas de gestación, de oligoamnios, con riñones macroscópicamente de apariencia y tamaño normal y Doppler uteroplacentario sin alteraciones1-2).
Se asocia a anuria fetal persistente e hipoplasia pulmonar secundaria, con otras anomalías, como las alteraciones en la osificación de los huesos del cráneo con agrandamiento de las fontanelas, conocido como hipocalvaria. Se han reportado múltiples casos con esta asociación, considerando la hipocalvaria como un resultado de la hipotensión mantenida generando hipoxia y alteración del crecimiento en los huesos del cráneo. Esta asociación es más conocida en los casos de madres que han recibido tratamiento con inhibidores de la enzima convertidora de angiotensina (IECA), pero se han reportado casos con esta asociación sin esta medicación, como es el caso que se presenta en esta revisión, lo que llevaría a plantear una similar fisiopatología3,4. Los elementos diagnósticos pre y posnatales se describen en la (Tabla 1).

En el caso clínico, el paciente presenta una insuficiencia renal mantenida, dada por anuria neonatal persistente, creatinina sérica de 3,26 y azoemia de 1,35 (valores normales de creatinina sérica en recién nacidos término: tres días de vida límite superior 0,5 mg/dl). El ionograma fue normal.
La mayoría de los pacientes fallece en el período neonatal, como es el caso presentado, por insuficiencia renal, hipotensión e hipoplasia pulmonar; sin embargo, hay casos reportados de sobrevivientes2-5).
La fisiopatología de la enfermedad no está aclarada, pero se origina en fetos con alguna de estas tres circunstancias2-7):
- Genética: enfermedad autosómica recesiva.
- Adquirida: exposiciones durante el embarazo a fármacos IECA6).
- En casos de gemelos con síndrome transfundido-transfusor.
El mecanismo patogénico común de las tres formas es la baja perfusión renal que se produce secundaria a la hipovolemia o hipotensión fetal (Figura 1). Dado que el desarrollo de los túbulos renales requiere una adecuada filtración glomerular, la disminución de ésta genera la alteración o ausencia en el desarrollo de los túbulos proximales2).
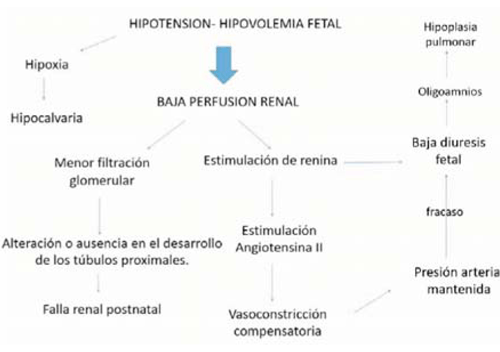
La baja perfusión renal genera la estimulación y liberación de renina, que produce angiotensina II, que tiene como función la vasoconstricción y el mantenimiento de la presión sanguínea sistémica. Este mecanismo compensatorio fracasa produciendo una disminución del flujo sanguíneo renal, generando una disminución de la diuresis fetal2-8).
La disgenesia tubular renal hereditaria es una enfermedad autosómica recesiva que obedece a mutaciones en los genes de cualquiera de los componentes del sistema renina-angiotensina-aldosterona (renina en cromosoma 1q32, angiotensina 1q42, enzima convertidora de angiotensina 17q23 y el receptor de angiotensina II tipo 1 3q24). Grivoubal9,10 realizó un reporte de más de 54 mutaciones en 48 familias no relacionadas. Las mutaciones más comunes, halladas en dos tercios de las familias (31 familias, 64,6%), son las producidas en el gen de la enzima convertidora de angiotensina con más de 33 mutaciones diferentes, algunas con relación de consanguinidad. En segundo lugar, son las mutaciones en el gen de la renina (20%), con más de diez mutaciones diferentes, siendo las menos comunes las mutaciones en el gen de angiotensina y en el receptor de angiotensina II, encontrados solo en cuatro y tres familias, respectivamente. La severidad del curso clínico no varía según la mutación hallada10).
La disgenesia tubular renal autosómica recesiva es genéticamente heterogénea, ya que se puede deber a diferentes tipos de mutaciones, distribuidos en cuatro genes del sistema renina-angiotensina-aldosterona. Todas estas mutaciones afectan la producción de renina y angiotensina (mutaciones en la enzima convertidora), o la eficacia de la angiotensina II, que es el producto final de la activación de la cascada. Esta vía común sugiere que la enfermedad no está específicamente vinculada al defecto de uno u otro gen, sino a la hipotensión sistémica y renal como consecuencia fisiológica de la inactivación del sistema renina-angiotensina-aldosterona. Esto demuestra el papel crucial de este sistema en el desarrollo renal.
Se han reportado casos de sobrevivientes con mutaciones en diferentes genes, un reporte de un caso con mutación en el gen de angiotensina5 y seis casos reportados en una revisión con tres mutaciones en el gen de la enzima convertidora de angiotensina, dos con alteraciones en el gen de angiotensina y un caso con mutación en el gen de la renina8. Esto apoya el planteo de que la severidad clínica no varía con la mutación, sino que es una consecuencia fisiopatológica de una vía común.
El uso de fármacos IECA produce un fenotipo de enfermedad muy similar al observado en la enfermedad autosómica recesiva, con disminución del flujo sanguíneo sistémico y renal y alteraciones en el eje renina-angiotensina-aldosterona. El mecanismo que plantea los antiinflamatorios no esteroideos durante el embarazo como posible causa es similar a lo expuesto anteriormente2,3,6,11).
En el caso de los fetos con síndrome transfundido-transfusor en embarazos monocoriales se produce en el feto transfusor una hipovolemia crónica que estimula el eje renina-angiotensina-aldosterona, con producción de angiotensina como vasoconstrictor, la cual disminuye el flujo renal con oliguria. La hipoperfusión y la hipofiltración glomerular alterarían el desarrollo de los túbulos proximales renales con el consiguiente oligoamnios2).
El diagnóstico definitivo se realiza con el estudio histológico de los riñones, en los cuales se hallan glomérulos agrupados y aumentados de tamaño, con túbulos con poco o nulo desarrollo formados por células pequeñas, pocos diferenciados, con apariencia “primitiva” (Figura 2) y (Figura 3). Esto indica un trastorno de la nefrogénesis1,2.
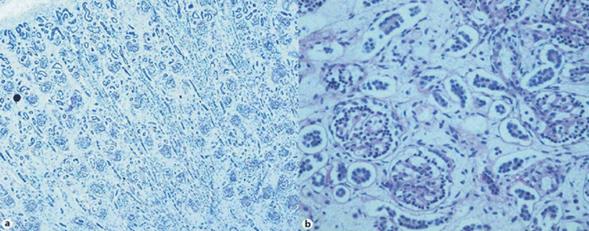
Figura 2: (a) Riñón normal. (b) Apiñamiento glomerular y túbulos inmaduros. Las superficies apicales de las células del túbulo proximal no han tomado la tinción de PAS(6).
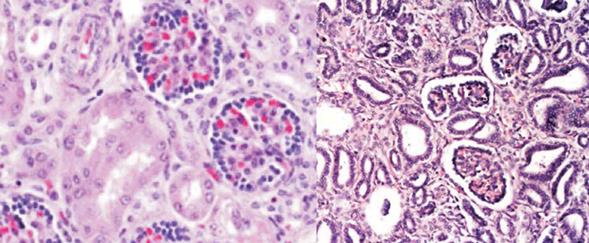
Figura 3: Corte histológico que muestra, abajo, túbulos inmaduros compatibles con disgenesia tubular renal, y, arriba, un riñón histológicamente normal para las 37 semanas (a) (imagen extraída de informe anatomopatológico del Centro Hospitalario Pereira Rossell de necropsia del paciente presentado).
Conclusiones
La disgenesia tubular renal es una enfermedad rara, subdiagnosticada, dado que no es considerada en muchos casos como diagnóstico diferencial y su diagnóstico definitivo se obtiene con el estudio histológico renal.
La realización de la necropsia es de suma importancia en recién nacidos fallecidos con sintomatología similar a la descrita para la realización del diagnóstico de la patología. En este caso no se obtuvo el resultado de la hibridación genómica, que hubiera sido de gran importancia para sellar el diagnóstico y realizar asesoramiento genético a la familia afectada.
Dado que presenta un 25% de riesgo de recurrencia familiar, el diagnóstico oportuno, junto con los avances en el origen de las mutaciones de los genes, ayudarán al asesoramiento genético a familias portadoras.

