











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Curriculum ScienTI
Curriculum ScienTI
 Permalink
Permalink
Introducción
La tos en la infancia es un síntoma muy frecuente y constituye uno de los motivos de consulta más comunes en la práctica pediátrica diaria. En la mayoría de los casos la causa son infecciones respiratorias banales, pero todo niño con tos que persiste más allá de las cuatro a ocho semanas se considera que tiene tos crónica y debe ser evaluado para descartar patologías específicas1. En caso de un síndrome canalicular exudativo persistente crónico y otitis recurrentes supuradas, debemos sospechar enfermedades respiratorias crónicas poco frecuentes que requieren una evaluación diagnóstica ampliada. Debemos estudiar fibrosis quística (FQ), inmunodeficiencias y discinesia ciliar primaria (DCP), entre otras. La DCP es una enfermedad de herencia principalmente autosómica recesiva, caracterizada por un defecto estructural de las células ciliadas presentes en el tejido respiratorio y gonadal, entre otros, que repercute en su función. Se han descrito más de 30 genes implicados, siendo el DNAH5 y DNAI1 los más ampliamente estudiados. No existe una única prueba diagnóstica gold standard para llegar al diagnóstico definitivo, siendo necesaria la existencia de un cuadro clínico compatible combinado con distintas pruebas diagnósticas (óxido nítrico nasal, análisis de la ultraestructura ciliar, función y batido ciliar, genética e inmunofluorescencia)2,3.
Se presenta el caso de un adolescente de 13 años con tos catarral y otitis crónica serosa desde los 6 meses de vida, realizándose el diagnóstico de DCP a los 9 años por la clínica, imagenología, estudio de microscopía electrónica de transmisión (MET) y genético.
Aspectos éticos: cuenta con el consentimiento informado del adolescente y sus padres, y la aprobación del Comité de Ética de la institución.
Caso clínico
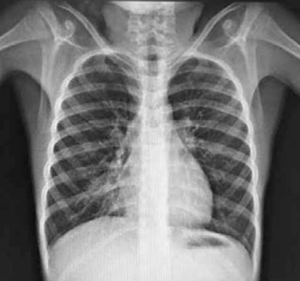
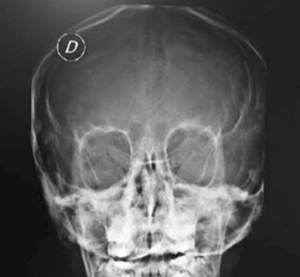
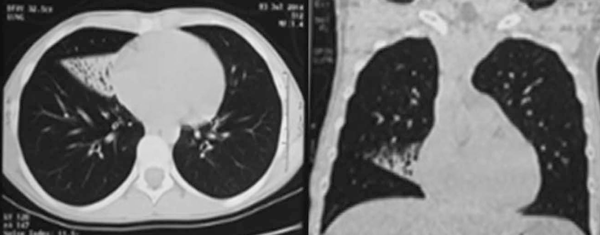
Adolescente de 13 años, sexo femenino, procedente de Montevideo, buen crecimiento y desarrollo, carné esquema de vacunación vigente, medio social, económico y cultural aceptable. Antecedentes familiares de padre y hermana con asma, abuela paterna con asma y aspergilosis broncopulmonar alérgica a los 50 años. La primera evaluación por neumología fue a los 9 años, se presenta con tos crónica catarral desde los 6 meses de vida, sin predominio estacional, no expectoración, drenaje postural, broncorrea, ni sinusitis. Se acompañaba de otitis crónica serosa que requirió colocación de tubos transtimpánicos a los 4 años, con persistencia de secreciones de oído medio, hipoacusia e impedanciometrías alteradas. Al examen se destaca: índice de masa corporal (IMC): 16,7 kg/m² (percentil 15); pleuropulmonar: frecuencia respiratoria 16, saturación de oxígeno ventilando espontáneamente al aire 98%, pico flujo espirado 350 L/m, moviliza secreciones con la tos, no remodelación de tórax, ni hipocratismo digital. No presentaba pólipos en la rinoscopía. Cardiovascular normal. En la radiografía de tórax se observa opacidad inhomogénea en lóbulo medio (Figura 1). La radiografía de senos faciales muestra opacidad de ambos senos maxilares (Figura 2). Con sospecha de bronquiectasias se solicitan estudios para descartar FQ, inmunodeficiencias, malformación congénita de la vía aérea y discinesia ciliar. Los resultados de tres test del sudor fueron normales: Cl 23 meEq/l (159 mg sudor), Cl 14 mEq/l (215 mg), Cl 19 mEq/l (180 mg), y la evaluación inmunológica fue normal. El exudado faríngeo no desarrolló gérmenes. La tomografía de tórax de alta resolución muestra múltiples bronquiectasias en lóbulo medio, disminuido de tamaño por atelectasia (Figura 3). Se realiza fibrobroncoscopía con lavado bronquioalveolar, en vistas de valorar la vía aérea, y valoración infecciosa. Se descarta lesión anatómica o dinámica en la vía aérea y cuerpo extraño, y el estudio bacteriológico y micológico fueron negativos. Con sospecha de DCP, se realiza biopsia de la mucosa bronquial y microscopía electrónica. Las muestras obtenidas fueron fijadas en glutaraldehído al 2,5% en tampón fosfato 0,1 M (pH: 7,2 - 7,4), y trasladadas a 4 grados al laboratorio de biología celular, Instituto de Investigaciones Biológicas Clemente Estable. Se observaron alteraciones ultraestructurales consistentes con DCP. Se realiza cepillado nasal de ambas fosas nasales, observadas vivas e incubadas a 37° al microscopio de contraste de fase en medio de cultivo médium 199 para determinar morfología, frecuencia y patrón de batido ciliar, constatándose movilidad ausente en todos los campos. La microscopía electrónica muestra ultraestructura ciliar con patrón 9+2, con ausencia de ambos brazos de dineína. El diagnóstico de funcionalidad con inmovilidad ciliar absoluta, cilios de ultraestructura con defecto de brazos internos y externos de dineína, confirma la DCP. Se solicita estudio genético molecular con secuenciación del gen DNAH5, que muestra una variante probablemente patológica con presencia en homocigosis de una duplicación c.10196dupT mutación frameshift p. Cys3400Metfsx54, no se encuentra en base de datos ni en la bibliografía consultada, y otra de significado incierto con presencia en heterocigosis de la variante c.A8387G: p.D2796G en el gen DNAH5. En la evolución, la paciente persiste con igual sintomatología, los cultivos de expectoración iniciales fueron negativos. A los 13 años el estudio bacteriológico de expectoración desarrolla Pseudomonas aeruginosa, se realiza tratamiento con gentamicina nebulizada y ciprofloxacina vía oral por 21 días. Los cultivos de control muestran desarrollo de Pseudomonas aeruginosa, por lo que se indica tobramicina inhalada por 28 días. Persiste desarrollo de abundantes colonias y se inicia colistina nebulizada cada 12 horas, inicialmente por tres meses, luego mensual alternando con tobramicina inhalada. De la evolución funcional, presentó espirometrías normales, con valores de volumen espiratorio forzado en el primer segundo (VEF1) estables de 95% del predicho, distancia recorrida en 6 minutos sin desaturación en la mar cha, ecocardiograma Doppler normal y audio metrías normales. En la tomografía de tórax, a los 13 años, persisten bronquiectasias asociadas a atelectasia en lóbulo medio, sin cambios significativos con respecto a la tomografía previa, hiperinsuflación compensadora del resto de los lóbulos; aisladas bronquiectasias en segmento basal medial de lóbulo inferior derecho, aso ciado a imágenes de árbol en brote. Agrega bronco rrea mucopurulenta 10 cc al día. Presentó adecuada adhe rencia al tratamiento con fisioterapia dos veces por día en domicilio, asistida por fisioterapeuta tres veces a la semana. El tratamiento antibiótico continuó con colistina nebulizada y cipro floxacina vía oral. Realiza ejer cicio y danza de forma regular.
Discusión
La DCP es un trastorno genéticamente heterogéneo, princi palmente autosómico recesivo, que se caracteriza por una disfunción de los cilios móviles2-5. Puede cursar con una amplia variedad de características clínicas que reflejan los numerosos órganos en los que los cilios son importantes para mantener el estado de salud3. Dado que los cilios móviles están presentes en todo el tracto respiratorio, cada pérdida de integridad estructural o funcional de los cilios conduce a un trastorno en el mecanismo de defensa innato primario del aclaramiento mucociliar4,6. Alrededor del 50% de los pacientes con DCP tienen situs inversus2,4,7 y se observa una reducción de la fertilidad en hombres y mujeres4,8. La prevalencia estimada de DCP es de 1 en 15.000-30.000 nacimientos vivos2,4, pero estudios recientes proponen que la prevalencia es mayor en poblaciones consanguíneas4. Se considera una enfermedad pulmonar rara, aunque se estima que su prevalencia entre los niños que padecen infecciones respiratorias recurrentes alcanza el 5%3.
Algunas características clínicas se superponen con otras afecciones, como la FQ, inmunodeficiencias, la aspiración pulmonar crónica, el asma y las infecciones virales respiratorias recurrentes5.
Se han identificado cuatro características clínicas claves9. La tos productiva diaria, todo el año; la rinosinusitis no estacional, diaria, que comienza a menudo inmediatamente después del nacimiento, y casi universalmente se presenta a los 6 meses de edad. Estos síntomas respiratorios pueden variar, pero nunca se resuelven por completo, incluso después de la terapia con antibióticos sistémicos. Aproximadamente el 80% de los niños con DCP tienen un historial de ser recién nacidos a término, con síndrome de dificultad respiratoria neonatal definido como la necesidad de oxígeno suplementario o soporte de ventilación con presión positiva durante más de 24 horas, sin una explicación clara. Aproximadamente el 40%-55% de los pacientes con DCP tienen defectos de lateralidad, como situs inversus totalis, mientras que otros defectos de lateralidad, como situs ambiguo, con o sin defectos cardíacos congénitos, se encuentran en aproximadamente el 12% de las personas afectadas3,9. Si las cuatro características clínicas claves están presentes, la sensibilidad y la especificidad son 21% y 99%, respectivamente. La otitis media crónica con derrame también es común, pero esta característica no distingue a los niños con DCP de los que no la tienen5,9. En recién nacidos a término, la combinación de situs inversus totalis y la insuficiencia respiratoria neonatal inexplicada es altamente sugestiva de DCP, incluso en lactantes que aún no han desarrollado síntomas respiratorios crónicos. Sin embargo, con dos de estas características claves, es poco probable que los pacientes tengan DCP, y las pruebas adicionales generalmente no están justificadas. Se deben considerar las pruebas de diagnóstico para DCP solo en aquellos pacientes que realmente se ajustan al fenotipo clínico5. Casi todos los pacientes presentan otitis media crónica de gravedad variable. En el momento del diagnóstico, la mayoría tiene antecedentes de perforaciones crónicas de membrana timpánica o han sido sometidos a timpanostomías con inserción de tubos de ventilación en membranas timpánicas. Muchos padecen pérdida conductiva de la audición durante la infancia, que mejora en la adolescencia3. Aunque esta carac terística no es pronóstica de DCP5,9, los hallazgos en el oído medio ayudan a diferenciar la DCP de FQ u otras causas de enfermedad pulmonar crónica3.
El diagnóstico a menudo se retrasa, incluso en niños con características clínicas clásicas, en parte relacionado esto con las limitaciones de las pruebas de diagnóstico disponibles5. Hoy en día, las características clínicas de la DCP son bien reconocidas, pero el diagnóstico sigue siendo difícil, especialmente cuando los pacientes presentan signos y síntomas inespecíficos, y cuando son necesarios equipos y pruebas de detección que incluyen mediciones de óxido nítrico (NO) nasal, pruebas de transporte mucociliar nasal y prueba de sacarina, que no están disponibles. Los cambios ciliares inespecíficos, que pueden ser inducidos por la exposición a contaminantes ambientales o infección, pueden parecer similares en la microscopía electrónica a los hallazgos obser vados en DCP5.
Los defectos ultraestructurales más frecuentemente hallados en la MET son en los brazos de dineína o en la disposición microtubular, o ambos10. Incluso la MET, que se consideraba como estándar de oro, no puede resolver el 30% de los pacientes porque la ausencia de defectos axonemales no excluye la DCP, dado que este porcentaje presenta una ultraestructura ciliar normal4,5,10).
Actualmente no existe una prueba estándar de oro para el diagnóstico y ninguna modalidad única tiene suficiente sensibilidad y especificidad diagnóstica4,5.
La DCP es causada por mutaciones patógenas bialélicas en uno de los muchos genes causantes identificados. Las limitaciones de la MET han hecho que las pruebas genéticas moleculares sean una alternativa atractiva, porque más del 50% de los pacientes con DCP poseen dos mutaciones patógenas en un gen causante de enfermedad conocido5,11). Debido a que las pruebas genéticas están cada vez más disponibles y los costos han disminuido, el impulso para considerar las pruebas genéticas moleculares como una prueba de diagnóstico de primera línea para DCP ha aumentado5. Las mutaciones bialélicas en el gen DNAH11 son una causa común de DCP, que representan aproximadamente el 22% de los casos, con resultados de ultraestructura ciliar por MET normal. Este gen codifica una proteína de cadena pesada componente del brazo de dineína externo axonemal del cilio10).
La tomografía de doble eje es una extensión de la técnica MET tradicional. Permite la visualización de estructuras ciliares en tres dimensiones (3D) que permiten una mejor resolución y comprensión de la ultraestructura ciliar10. Un estudio publicado recientemente demuestra que las mutaciones de DNAH11 resultan en una anormalidad característica de la ultraestructura ciliar detectable por tomografía electrónica, pero no por MET tradicional10.
En pacientes que presentan un fenotipo clínico altamente sugestivo, la Sociedad Americana de Tórax (ATS) recomienda usar un panel genético extendido como prueba diagnóstica antes que el análisis de MET o pruebas genéticas estándar (<12 genes)9.
Por lo tanto, la implementación del diagnóstico genético y molecular es más bien una necesidad, independientemente de si es una confirmación de un diagnóstico clínico o una sospecha de DCP4. Recientemente, la disponibilidad de técnicas de secuenciación de alto rendimiento contribuyó a la identificación rápida de nuevos genes causantes de DCP, lo que resultó en la identificación de más de 40 genes hasta el momento, lo que permitió que se describiera genéticamente el 65% de los casos4.
Los procedimientos diagnósticos de DCP son complejos, requieren una infraestructura costosa y un equipo experimentado de médicos, genetistas y microscopistas. El diagnóstico molecular es una herramienta útil especialmente en enfermedades con presentación clínica superpuesta. Sin embargo, debido a la heterogeneidad genética de la DCP, aproximadamente el 35% de los casos aún carecen de confirmación de la base genética de la enfermedad4. La prueba de panel genético extendido tiene limitaciones clínicas. Los resultados negativos del panel extendido no excluyen la DCP, porque es probable que aún se descubran algunos genes adicionales5.
Las variantes de significado desconocido pueden proporcionar resultados no diagnósticos y la interpretación incorrecta de los resultados de las pruebas genéticas puede dar lugar a diagnósticos falsos positivos o falsos negativos5.
La ATS sugiere que en pacientes cooperativos de 5 años o mayores, con un fenotipo clínico consistente con DCP y con FQ excluido, realizar inicialmente la prueba de NO nasal antes que MET o pruebas genéticas. Debido a que los valores de NO nasal pueden disminuir transitoriamente con infecciones respiratorias virales agudas o sinusitis, está indicado establecerlo en dos ocasiones separadas. En pacientes con un fenotipo clínico compatible y NO nasal bajo en dos ocasiones, se puede establecer un diagnóstico presuntivo de DCP. La prueba de NO nasal no es invasiva, es relativamente de bajo costo y proporciona resultados inmediatos. Sin embargo, hay limitaciones: se realiza en centros especializados y la falta de estándares para niños menores de 5 años. Incluso con medidas bajas de NO, los pacientes deberían continuar con estudios diagnósticos incluidos genéticos o pruebas de MET, que pueden proporcionar información pronóstica a largo plazo, planificación familiar y futuras tera pias específicas de mutación9.
La ATS sugiere no utilizar el análisis del movimiento ciliar por videomicroscopía digital de alta velocidad como prueba diagnóstica única en pacientes con alta probabilidad de tener DCP, dado que requiere una experiencia sustancial y se realiza mejor en centros especializados en DCP. Movimientos lentos o anormales, como ocurre después de una lesión o infección de las vías respiratorias, pueden ser un defecto adquirido y conducir a una conclusión positiva falsa. También puede verse afectado por la manipulación de muestras de tejido fresco y puede llevar a un fenotipo funcional diferente9.
El algoritmo diagnóstico propuesto por la ATS, si se sospecha el diagnóstico, con por lo menos dos de las cuatro características claves, es realizar inicialmente test de NO nasal (con dispositivo de quimioluminiscencia y protocolo estandarizado en mayores de 5 años). Si es bajo, se hace diagnóstico (luego de descartar FQ), igualmente realizar pruebas adicionales con paneles genéticos extendidos (primera línea), y MET de ultraestructura ciliar. Si es normal y existe fuerte sospecha, realizar pruebas genéticas. Si no es posible realizar pruebas de NO, realizar panel genético extendido. Si muestra variantes patogénicas bialélicas de gen asociado a DCP, se hace diagnóstico. Si muestra una variante patogénica única o ninguna, realizar microscopía electrónica de ultraestructura ciliar. Si muestra un defecto reconocido, se confirma DCP, y si es normal, el diagnóstico aún es posible9).
Finalmente, el análisis de los antecedentes genéticos de la DCP conducirá a una mejor comprensión de las enfermedades y posiblemente al diseño de una nueva terapia dirigida molecularmente4.
Destacamos que la paciente en la evolución presentó estudios de función pulmonar normales, con VEF1 mantenido a pesar de presentar una infección crónica por Pseudomona aeruginosa, con aumento del daño estructural de la vía aérea. Las infecciones recurrentes y crónicas, y la inflamación persistente del sistema respiratorio, resultan en daño pulmonar progresivo12. En un estudio multicéntrico realizado en Israel y en Europa en pacientes con DCP, se demostró que la prevalencia de colonización con Pseudomona aeruginosa aumenta con la edad, y esto se asoció con una función pulmonar más baja y puntajes tomográficos más severos; sin embargo, la tasa de disminución del VEF1 durante el período de estudio entre los grupos colonizados y no colonizados fueron similares. Otros trabajos en niños y adolescentes no encontraron correlación entre la infección por Pseudomona aeruginosa y VEF112. Esto apunta a la necesidad de realizar estudios para determinar la erra dicación de Pseudomona aeruginosa.
Concluimos que si bien el diagnóstico es complejo y es una enfermedad subdiagnosticada, ante la aparición de los síntomas y signos característicos, debemos de sospechar la DCP para diagnosticarla precozmente y brindar una mejorar calidad de vida de estos pacientes.

