











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Introducción
El hiperinsulinismo congénito (HC) es una alteración del metabolismo de la glucosa que se debe a múltiples etiologías. La manifestación común es la presencia de hipoglucemia persistente de difícil control que se asocia con valores inapropiadamente elevados de insulina para los valores de glucosa observados1. Es la causa más frecuente de hipoglucemias reiteradas y severas en la etapa neonatal. Presenta un riesgo elevado de alteración profunda del neurodesarrollo a lo largo de la vida2. La incidencia mundial es de 1 cada 50.000 nacidos vivos3. En Uruguay se desconoce su frecuencia, en esta revisión se pudo documentar un caso publicado4.
Puede ser transitorio debido a factores de riesgo como asfixia perinatal, retraso del crecimiento intrauterino, diabetes mellitus materna, o estar asociado a síndromes de sobrecrecimiento, como Beckwith-Wiedemann5. Las formas genéticas de HC se deben a una mutación en los genes implicados en la regulación de la secreción de insulina5. Avances actuales en biología molecular y genética lograron identificar hasta el momento 12 genes, que al estar afectados pueden dar lugar a la expresión de la enfermedad. Estos avances permitieron contar con opciones terapéuticas específicas y eficaces5,6. Sin embargo, la incidencia de daño neurológico permanece prácticamente incambiada2. El rápido reconocimiento, diagnóstico y tratamiento es fundamental para evitar las secuelas neurológicas vinculadas a la hipoglucemia. Los recién nacidos, diagnosticados tempranamente y tratados adecuadamente, pueden tener una vida plena.
A continuación se presenta el caso clínico de una paciente con hipoglicemias graves de difícil control desde el nacimiento, secundarias a hiperinsulinismo congénito de tipo difuso, que presenta una evolución fatal.
Caso clínico
Madre, 33 años. Sana. No consanguinidad. Cursó tercera gestación de la misma pareja, dos hijos sanos. Embarazo controlado. Rutinas obstétricas normales durante el embarazo. Inicio espontáneo de trabajo de parto, rotura de membranas de 6 horas, parto vaginal en cefálica. Recién nacido de sexo femenino, 35 semanas, vigoroso (Apgar 8/9), peso al nacer: 2.965 g (score Z + 1,7), grande para la edad gestacional. Índice ponderal: 2,6. Longitud: 48 cm (score Z + 1), perímetro craneano: 31,5 cm (score Z: -0,6). Gasometría de cordón sin acidosis. Examen físico normal al nacer. A las 33 horas de vida, paciente temblorosa con quejido intermitente, glicemia: 28 mg/dl. Se inicia aporte de glucosa intravenosa a 6 mg/kg/min, con aumentos progresivos hasta 16 mg/k/min. Debido a la reiteración de valores de glicemia capilar menores a 0,40 g/l, con un aporte de glucosa superior a 12 mg/kg/min, se inicia tratamiento con hidrocortisona y se solicita muestra crítica cuyos resultados son: insulinemia 71 mUI/l (valores normales: 2,6 a 11), cortisol 27,3 ug/dl (valores normales 6,2 a 19,4). Cuerpos cetónicos negativos. Ácidos grasos libres en sangre: 0,55 (0,09-0,6). Amonio: 112 ng/dl (90-150).
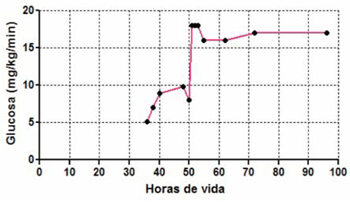
Se realiza diagnóstico clínico de hiperinsulinismo congénito y se inicia tratamiento con diazóxido. Persiste con altos requerimientos de glucosa i/v y enteral (Figura 1) sin respuesta al diazóxido con dosis de 30 mg/k/día.
Tomografía con emisión de positrones (PET) con 686a DOTATATE normal.
Estudio genético realizado en la Universidad Exeter, Londres, mostró dos alelos mutados del gen ABCC8, consistente con el diagnóstico de hiperinsulinismo congénito autosómico recesivo.
Evolución
Debido a la alteración de la succión deglución se alimentó inicialmente por sonda nasogástrica, en la evolución con sonda nasoyeyunal, realizándose a los tres meses gastrostomía percutánea. Resistencia al tratamiento con diazóxido. Se inició tratamiento con octeotride, con buena respuesta inicial, pero el paciente continuó requiriendo aporte intravenoso de glucosa para mantener glicemias normales (Figura 2).
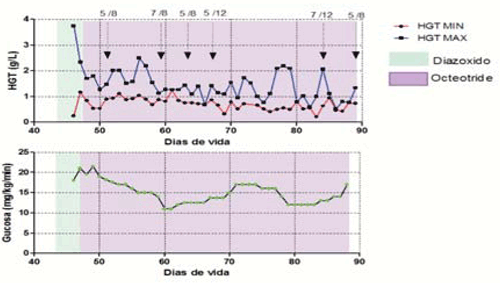
A los cuatro meses de vida, debido a falta de respuesta al tratamiento médico, se realiza pancreatectomía subtotal. En el posoperatorio presenta hiperglicemias de difícil manejo que requieren infusión de insulina, sin lograrse estabilidad metabólica. Fallece a los seis meses de vida cursando sepsis a Candida.
Discusión
Durante la vida intrauterina el feto recibe glucosa a través de un mecanismo de difusión facilitado a través de la placenta. Luego del nacimiento, en recién nacidos de término sanos, sin factores de riesgo para hipoglucemia, los niveles de glucosa en plasma tienden a mostrar una aguda disminución durante las primeras 24-48 horas, para luego normalizarse5. No existe unanimidad acerca de los valores de glicemia normales en las primeras 24 horas de vida, pero existe consenso que luego del segundo día de vida deben mantenerse valores superiores a 50 mg/dl7. En el recién nacido la presencia de convulsiones, temblores, irritabilidad, cianosis, apnea, letargia y dificultad en la alimentación pueden ser síntomas de hipoglicemias. Cuando la hipoglicemia es transitoria, el 90% de los casos es asintomático, pero en el caso de hipoglicemia persistente la presencia de manifestaciones clínicas es la regla7. Se define como hipoglicemia transitoria si se limita a los primeros 5-7 días de vida, e hipoglicemia persistente si se mantiene más allá de la semana de vida. La causa más frecuente de hipoglicemia persistente es el hiperinsulinismo congé nito8.
A las 33 horas de vida la paciente se presenta temblorosa, con quejido intermitente; se reitera glicemia, siendo de 28mg/dl. Por presentar una hipoglicemia sintomática, se ingresa a sector de internación para alimentación y aporte de glucosa intravenosa. Situaciones clínicas como la presentada, que requieren elevado aporte de glucosa intravenosa de 16 mg/kg/min para mantener la glicemia normal, deben conducir a la valoración diagnóstica para HC. Se obtiene muestra de sangre crítica en el momento en que la paciente presenta hipoglucemia de 38 mg/dl. La muestra crítica se usa para medir las concentraciones plasmáticas de las hormonas y combustibles alternativos involucrados en la respuesta fisiológica al ayuno. Para minimizar los resultados falsos positivos, el umbral de glucosa en plasma para obtener una muestra crítica debe ser menor de 50 mg/dL8. La célula beta pancreática regula la secreción de insulina por intermedio de canales de potasio sensibles a ATP (canal KATP). Estos canales son un complejo octamérico, conformados por cuatro subunidades rectificadoras de potasio (Kir6.2) que forman el núcleo del canal y cuatro subunidades del receptor de alta afinidad de sulfonilurea (SUR1) que ejercen una función regulatoria9,10.
Las anomalías en 12 genes han sido implicadas en HC: mutaciones de los canales KATP o canalopatías, y desórdenes enzimáticos que favorecen un incremento en la relación ATP/ADP intracitoplasmático o metabolopatías2,6-11.
Los primeros que podríamos definir como canalopatías, o defectos del canal KATP, se presentan por mutaciones en los genes KCNJ11 y ABCC8, que están localizados en el cromosoma 11p15.1 que codifican para las subunidades Kir6.2 y SUR1, respectivamente12. Hasta la fecha se han descripto 150 mutaciones inactivadoras de ABCC8 y 24 de KCNJ11, las cuales desde el punto de vista clínico se comportan como refractarias al manejo con diazóxido13.
Los trastornos más comunes son los que afectan a los genes de los canales KATP y son predominantemente mutaciones recesivas14.
La presentación de la enfermedad es variable, siendo la forma más grave y frecuente la que se presenta en el período neonatal, como en el caso señalado. Debe considerarse esta patología en recién nacidos macrosómicos o GEG con una hipoglicemia persistente15.
Según la histopatología pancreática, el HC se puede clasificar en tres tipos de causas moleculares diferentes: el HC focal (HCF), el difuso (HCD) y el atípico. El HCF se caracteriza por la presencia de uno o varios focos de proliferación de las células beta pancreáticas hipersecretoras de insulina, mientras que la forma HCD ocurre cuando todo el islote de células en el páncreas son anormales16. En la mayoría de los casos, el HCD es heredado de forma autosómica recesiva, produciéndose por dos mutaciones en los genes ABCC8/KCNJ11, una en cada alelo, como es el caso de nuestra paciente, mientras que el HCF en el 40%-60% de los casos es esporádico3-5. Cuando los casos son difíciles de categorizar histológicamente, se clasifican como formas atípicas17.
El grupo de desórdenes en HC es amplio, por lo cual es clave un adecuado abordaje diagnóstico. Una aproximación primaria puede basarse en los criterios bioquímicos de Aynsle-Green y colaboradores11 (Tabla 1) y debe realizarse en base a la obtención de una muestra crítica de sangre obtenida durante un episodio espontáneo o provocado de hipoglucemia. La respuesta glucémica al glucagón debería ser evaluada18).

En el caso mencionado, la paciente cumplía con los criterios bioquímicos diagnósticos, dado que presentaba requerimiento de glucosa mayores a 8 mg/kg/min, acom pañado de elevación de insulina durante el episodio de hipoglicemia, con cuerpos cetónicos bajos. Sin embargo, un error común en el diagnóstico de HC es que la concentración de insulina no siempre es elevada al momento de la hipoglucemia. El hecho de que los niveles de insulina no se encuentren aumentados al momento de la hipoglucemia puede deberse a la liberación periódica de insulina, que se pierde en una sola muestra, o a la rápida depuración hepática, de tal manera que el hígado es expuesto a altos niveles de insulina y no se ve reflejado en la sangre venosa periférica. Esto también podría ser debido a la actividad de enzimas degradadoras de insulina que se encuentran presentes en muestras hemolizadas19. Por lo tanto, el diagnóstico de HC con frecuencia debe basarse en otra evidencia de la acción excesiva de la insulina, tales como la supresión en los niveles de ácidos grasos libres y cuerpos cetónicos. El cortisol se encuentra elevado debido a una adecuada respuesta de las hormonas contrarreguladoras, habitualmente por arriba de 20 mcg/dl (500 nmol/L). Este último hallazgo puede ser de utilidad cuando el diagnóstico está en duda o es difícil. El panhipopituitarismo neonatal puede tener un perfil bioquímico idéntico al HC. Si el cortisol y los niveles de la hormona del crecimiento de la muestra crítica no son elevados, las pruebas de estimulación apropiadas sirven para evaluar hipopituitarismo20. Es posible encontrar bajos niveles de cortisol en relación con hipoglicemia en neonatos con HC, correspondiendo a una respuesta inadecuada transitoria del eje hipotálamo hipofisiario para producir ACTH21.
El perfil bioquímico que presenta la paciente es compatible con el diagnóstico de HC, con excepción del valor de ácidos grasos libres normales, habitualmente éstos se encuentran disminuidos, como se mencionó previa mente.
En cuanto al abordaje terapéutico, será muy diferente según el tipo de HC que padezca el paciente, de modo que es crucial establecer una correcta clasificación genética e histopatológica para mejorar tanto el tratamiento como el pronóstico de estos niños. Las formas dominantes, además de ser menos graves, responden muy bien al tratamiento con diazóxido y no suele ser necesaria la cirugía. En cambio, la forma recesiva asociada a los genes ABCC8/KCNJ11, como el caso de nuestra paciente, a menudo no responde, o lo hace en forma muy limitada al tratamiento farmacológico. En cualquiera de los casos la finalidad del tratamiento es prevenir el daño neurológico debido a la hipoglicemia, establecer un patrón normalizado de alimentación respecto al contenido, el volumen y la frecuencia de las comidas y mantener la integridad de la familia, ya que esta enfermedad puede tener una gran repercusión sobre la convivencia2,3-16).
El cerebro del recién nacido tiene una tasa más alta de consumo de glucosa en comparación con los adultos, por lo tanto es más vulnerable a la lesión cerebral causada por la hipoglicemia. Además, se priva al cerebro de la producción de cuerpos cetónicos, como se señaló previamente, que son combustibles alternativos cruciales22. Por lo tanto, la hipoglucemia grave y recurrente resultante del HC, si no se trata, se asocia con daño cerebral irreversible. La frecuencia de retrasos del neurodesarrollo en el HC es alta, 30% a 50%, y, lo que es más importante, esto afecta no solo a los niños con formas congénitas y permanentes de HC, sino también a los niños con reconocimiento transitorio de HC23.
El objetivo del tratamiento es mantener la glucosa en plasma mayor a 70 mg/dL, incluso durante los períodos de ayuno8.
El plan alimentario no debe ser considerado exitoso si no se cumplen los objetivos mencionados. En la paciente la alimentación inicial fue en base a SNG y SNY, ocurriendo efectos no deseados de la alimentación forzada, como ser el reflujo gastroesofágico, requiriendo en la evolución una gastrostomía para un mejor manejo del alto aporte calórico24.
El pilar de la terapia farmacológica es el diazóxido, un fármaco de similares características al grupo de tiazidas pero que carece de efecto diurético, actúa como hiperglucemiante al inhibir la secreción de insulina actuando sobre el canal K+ dependiente. El diazóxido generalmente es efectivo en todas las formas de HC donde la función del canal KATP está intacta, pero las mutaciones del canal KATP pueden no responder al mismo2. En estos casos se recomienda comenzar rápidamente con un ensayo de diazóxido a dosis máximas durante cinco días (15 mg/kg/día). La falta de respuesta al diazóxido se mide de acuerdo a las dosis máximas utilizadas y a la necesidad de altos aportes de glucosa. Nos permite catalogar al HC como no respondedor y nos sugiere la probable localización del defecto en los canales K+ dependientes. La respuesta al diazóxido debe ser examinada después de cinco días en dosis máximas (3-5 vidas medias). La falta de respuesta indica que el pa ciente puede ser candidato a cirugía17.
Dentro de los múltiples efectos adversos del diazóxido, el más severo es la retención de líquidos. En el recién nacido generalmente se administra junto al diazóxido un diurético tiazídico para evitar esta complicación25.
Debido a la falta de respuesta al diazóxido por la mutación de base, se inició octreotide, un análogo de la somatostatina. La primera respuesta a la administración de octreotide es generalmente hiperglucemica con taquifilaxia después de 48 horas, que requiere un ajuste de las dosis26. Comenzar con dosis bajas de octeotride puede evitar esta complicación27. Un análogo de somatostatina de acción prolongada, lanreotida, se ha usado con éxito en niños, aunque las limitaciones de dosificación restringen su uso28. Una vez instaurado el tratamiento inmediato adecuado, el siguiente objetivo debería ser la clasificación del tipo de hiperinsulinismo, tanto desde el punto de vista molecular como anatomopatológico.
Antes de la cirugía, el análisis de la mutación genética de los genes del canal KATP (ABCC8 y KCNJ11) puede proporcionar información útil para la diferenciación entre las formas focal y difusa. La presentación neonatal, la gravedad (ambas estrechamente relacionadas) y la respuesta al tratamiento con diazóxido serán sugestivas de hiperinsulinismo asociado a ABCC8/KCNJ113). Los pacientes con una mutación heterocigota recesiva o compuesto homocigotos en los genes ABCC8 o KCNJ11 presentan HC de tipo difuso, como el de la paciente expuesta. En dicha paciente se encontraron dos alelos mutados del gen ABCC8. Estos pacientes suelen ser los que no responden al tratamiento médico y corresponden al 60%-70% de los casos de hiperinsulinismo. Si no se puede acceder al estudio molecular, la tomografía por emisión de positrones (PET) con 18F-Fluoro-L-Dopa como marcador permite diferenciar la forma difusa de la localizada, con sensibilidad de 88% a 94% y especificidad cercana al 100%29. La acumulación anormal del 18F-Fluoro-L-Dopa en el páncreas orienta a HCF, pero los pacientes con HCD presentan una distribución difusa del marcador30. Como en Uruguay, al momento de asistir el caso clínico que se comunica, no se pudo acceder al marcador, se realizó la técnica con DOTATATE, con conocimiento de sus limitaciones. El tratamiento quirúrgico está indicado en niños que no pueden manejarse médicamente o que pueden tener una lesión focal de hiperinsulinismo que puede ser curado quirúrgicamente. En los casos de HCD, como el que presentamos, es recomendable intentar inicialmente un tratamiento conservador para evitar la cirugía. Los pacientes que requieren intervención quirúrgica se someten a una pancreatectomía casi total con colocación de tubo de gastrostomía. El tubo de gastrostomía es necesario para el manejo del posoperatorio porque la mayoría de los pacientes continúan teniendo hipoglucemia, aunque menos grave17. Para los niños con enfermedad difusa, la decisión de proceder con la cirugía es compleja y requiere una cuidadosa consideración de los riesgos y beneficios. Es fundamental contar con la presencia de un cirujano y anatomopatólogo con experiencia en la cirugía pancreática pediátrica31. Una vez realizada la pancreatetomía, no es infrecuente que ésta resulte ineficaz y se precise continuar con tratamiento médico (5%-25% de los casos)32. Las complicaciones que pueden presentar los pacientes sometidos a dicha operación son la diabetes mellitus, insuficiencia pancreática exocrina, problemas de alimentación y más episodios de hipoglicemia. El caso señalado presentó como complicación una diabetes de difícil manejo, con hiperglicemias mantenidas, las cuales pueden ocasionar disfunción inmunológica. La paciente fallece a los 6 meses de vida en el contexto de una sepsis a Candida.
En el año 2014, sirolimus, un agente inmunosupresor con capacidad antiproliferativa, se informó como un tratamiento novedoso, pero los estudios posteriores no han demostrado su eficacia33. Teniendo en cuenta los riesgos de una mayor susceptibilidad a infecciones severas asociadas con sirolimus, su uso no se recomienda actualmente34. Algunos autores recomiendan una dieta cetogénica como tratamiento coadyuvante, aunque este tratamiento requiere una mayor investigación. Los hallazgos iniciales sugieren que una dieta cetogénica podría tener un efecto neuroprotector en casos seleccionados35. Entre los fármacos prometedores para el tratamiento del HC se encuentra la exendina GLP-1, un antagonista del receptor del péptido 1 similar al glucagón. Dada la falta de terapias disponibles, el antagonismo del receptor GLP-1 representa un nuevo objetivo terapéutico para controlar la hipoglucemia en el HC y podría tener un impacto beneficioso sobre la morbilidad y me jorar los resultados a largo plazo para este trastorno devasta dor36.
El manejo del HC requiere un enfoque multidisciplinario que incluya a pediatras, endocrinólogos, imagenólogos, anatomopatólogos y cirujanos.

