











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La osteogénesis imperfecta engloba un grupo heterogéneo de enfermedades hereditarias del tejido conectivo, caracterizadas por una disminución de la masa ósea y una susceptibilidad aumentada a las fracturas1.
Tiene una incidencia general de 1 en 15.000-20.000 nacimientos2.
La osteogénesis imperfecta es causada por defecto en la síntesis o el metabolismo del colágeno tipo I. El colágeno tipo I está compuesto por dos cadenas pro a1 y pro a2, codificadas por los genes COL1A1 (cromosoma 17) y COL1A2 (cromosoma 7), respectivamente1,3.
La mayoría de los casos son de herencia autosómica dominante y corresponden a mutaciones en los genes COL1A1 y COL1A2, responsables de la codificación de las cadenas a1 y a2 del colágeno tipo I. Las formas autosómicas recesivas corresponden a mutaciones en genes que codifican las proteínas involucradas en la modificación postranscripcional del colágeno tipo I2,4.
Esta enfermedad se caracteriza por una amplia gama de formas de presentación, desde formas leves que se manifiestan como osteoporosis prematura o pérdida mineral ósea severa en la posmenopausia, hasta formas letales que causan la muerte en el período neonatal1.
Caso clínico
Madre de 23 años, sana. Niega consanguinidad y casos de similares características en la familia. Presenta como antecedente obstétrico cinco gestaciones previas que finalizaron en abortos espontáneos, se desconoce la causa subyacente. Cursa su sexta gestación, con un embarazo de captación precoz a las 12 semanas, bien controlado en cantidad y calidad con diagnóstico prenatal de probable displasia esquelética. Este diagnóstico presuntivo está basado en ecografías obstétricas morfoestructurales realizadas a las 22 y 24 semanas de gestación que muestran: acortamiento y curvatura de los huesos largos, todos por debajo del percentil 5, relación toraco-abdominal normal, elementos de hipoplasia pulmonar leve, braquicefalia con estructuras intracraneanas dentro de límites normales, con un ecocardiograma fetal que es normal.
Inicia espontáneamente el trabajo de parto, se realiza una operación cesárea por presentación en podálica. La rotura de membranas ovulares es intracesárea con líquido amniótico meconial fluido.
Se obtiene una recién nacida de sexo femenino, de 40 semanas de edad gestacional, con un peso al nacimiento de 2.495 g, una longitud de 39 cm y un perímetro cefálico de 34,5 cm. Apgar 5/8. Sin acidosis de cordón. Corresponde a un bajo peso al nacimiento, siendo pequeña para su edad gestacional, asimétrica a expensas de la talla, con un score Z para el peso de -2,0; para la longitud Z de -4,9, con perímetro cefálico adecuado para su edad gestacional.
Del examen físico al nacimiento se destacan múltiples alteraciones óseas: tórax pequeño, acortamiento y curvatura de los cuatro miembros, manos y pies pequeños, ausencia casi total de la calota craneal y paladar ojival.
Desde el nacimiento presenta dificultad respiratoria progresiva, requiriendo intubación orotraqueal y asistencia ventilatoria mecánica.
Se realizaron radiografías que muestran: huesos largos acortados, deformados, con angulaciones y arcos característicos de las múltiples fracturas intrauterinas (fracturas en acordeón) (Figura 1); en tórax presenta múltiples fracturas costales, con el aspecto de monedas apiladas con una caja torácica pequeña con poco volumen pulmonar (Figura 2). En huesos faciales y craneanos se observa marcada deficiencia de la osificación (Figura 3).
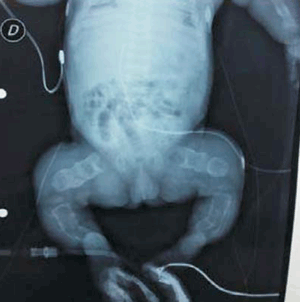
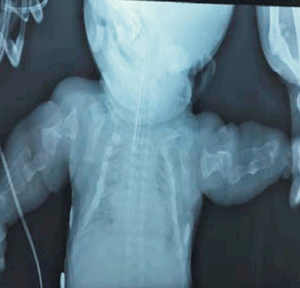
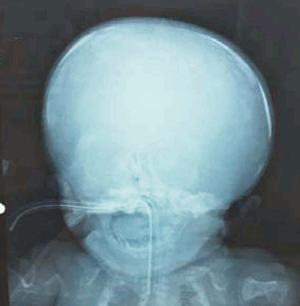
Se plantea, en base a la clínica y los hallazgos radiológicos, el diagnóstico de osteogénesis imperfecta neonatal severa o tipo II, se solicita secuenciación completa de las regiones codificantes y zonas intrónicas adyacentes de los genes COL1A1 y COL2A2.
Se realiza el estudio genético que informa variante c.2152G>A (p.Gly718Ser) en heterocigosis en el gen COL1A2. Esta variante ha sido descrita como variante probablemente patogénica asociada a osteogénesis imperfecta tipo II.
La paciente fallece a los 11 días de vida por insuficiencia ventilorrespiratoria progresiva severa.
En el estudio necrópsico se observan miembros cortos simétricos, hipocalvaria, fracturas múltiples en distintas etapas evolutivas; en la histología presenta osificación intramembranosa deficiente; hallazgos compatibles con el diagnóstico de osteogénesis imperfecta.
Discusión
Con este caso clínico se pretende mostrar una de las formas más severas de presentación de la osteogénesis imperfecta, la importancia del diagnóstico prenatal y el asesoramiento genético. Recordando que se trata de una madre joven y presenta como antecedente cinco gestaciones previas que culminaron en abortos espontáneos, sin causa aclarada.
En la paciente se encontró una mutación del gen COL1A2 (p.Gly718Ser) heterocigota, variante que ha sido previamente descrita en las bases de datos Human Gene Mutation Database (HGMD), en la cual se cita una sola referencia en relación con la osteogénesis imperfecta tipo II5,6. No existen casos descritos en otras bases de datos como Leiden OPEN Variant Database (LOVD) y ClinVar. Por lo tanto, se describe esta mutación como una variante probablemente patogénica asociada a la osteogénesis imperfecta tipo II5-8.
La prevalencia estimada de todos los tipos combinados de osteogénesis imperfecta es de 0,5 en 10.000 nacimientos9.
La mayoría de los individuos con osteogénesis imperfecta (>90%) presentan mutación de los genes COL1A1 y COL1A2. Las formas leves presentan mutaciones que resultan en defectos cuantitativos del colágeno tipo I. Mientras que en las formas severas se afecta la conservación de la glicina (los residuos de glicina son esenciales para la organización de la triple hélice de las proteínas del colágeno), resultando en un colágeno cualitativamente anormal, como parece ser el caso de nuestra paciente1,10.
En la mayoría de los casos las mutaciones de los genes COL1A1 y COL1A2 son de herencia autosómica dominante1. Si bien las formas recesivas suelen ser más severas, también pueden serlo las formas dominantes dependiendo de la mutación.
En los casos autosómicos dominantes la enfermedad a menudo es heredada de alguno de los padres. Si bien la mutación es la misma, la expresión de la enfermedad puede variar entre individuos de la misma familia en cantidad de fracturas o severidad. El afectado tiene un riesgo de transmisión de la enfermedad de 50% para cada embarazo.
Son comunes las mutaciones espontáneas de novo y explican los casos de osteogénesis imperfecta que son autosómicas dominantes en niños nacidos de padres sanos. En estos casos se sugiere evaluación clínica completa, estudio genético y eventualmente radiológico de los padres para asegurar que se trata de un caso esporádico.
Existe mosaicismo gonadal en 3%-5% de los casos; esto significa que la mutación podría estar presente en las células reproductivas (gametos) de alguno de los progenitores, siendo ellos asintomáticos, y explicaría la recurrencia en otro hijo afectado. En estos casos se estima un riesgo de repetición de 5% para próximos hijos de la pareja11.
En este caso clínico se plantea por frecuencia la mutación de novo; no pudiéndose descartar el mosaicismo gonadal dado que los padres son fenotípicamente sanos, pero con el antecedente de cinco abortos espontáneos previos, cuya causa pudo haber tenido como base esta mutación. Aquí cobra relevancia el asesoramiento genético de los progenitores en vistas a futuros embarazos. Este estudio sería, en este caso, para confirmar que la variante no está presente en el genoma constitucional de los padres (de ser así, y siendo estos sanos, dudaríamos de la patogenicidad de la variante), y eventualmente usando métodos de next generation sequencing, se pueden encontrar evidencias de mosaicismo gonadal si se evidencia la presencia de la variante en una baja frecuencia de lecturas.
La clasificación inicial fue la de Sillence; las dividía en cuatro categorías, basado en las características clínicas, la severidad de la enfermedad y los hallazgos radiológicos2,11.
Posteriormente, el Grupo de Nomenclatura Internacional para Trastornos Constitucionales ICHG del Esqueleto (INCDS) tomó la decisión de incluir cuatro categorías adicionales, englobando en estas ocho categorías los genes involucrados10.
- Tipo I es una forma de presentación leve, se caracteriza por la presencia de escleróticas azules, osteopenia generalizada y pérdida progresiva de la audición. Genes involucrados COL1A1 y COL1A2.
- Tipo II presenta una fragilidad ósea extrema, que conduce a la muerte en el período perinatal frecuentemente por fallo ventilatorio. Genes involucrados COL1A1 y COL1A2.
- Tipo III se caracteriza por ser gravemente deformante dada la fragilidad ósea intensa; retraso del crecimiento. Genes involucrados COL1A1 y COL1A2.
- Tipo IV, la severidad es intermedia entre la tipo I y la tipo III. El pronóstico en general es bueno. Genes involucrados COL1A1 y COL1A2.
- Tipo V, osteogénesis imperfecta con calcificación de membranas intraóseas. Moderadamente deformante. Gen involucrado IFITM5.
- Tipo VI de moderada a gravemente deformante. Genes involucrados IFITM5, SERPINF1.
- Tipo VII moderadamente deformante. Gen involucrado CRTAP.
- Tipo VIII de moderada a gravemente deformante. Gen involucrado LEPRE1.
La osteogénesis imperfecta tipo II presenta fracturas o arqueamientos de huesos largos con remodelación que conduce a huesos largos arrugados (similares a un acordeón). Costillas gruesas continuamente moldeadas debido a múltiples sitios de fractura o costillas finas (descrito anteriormente, según Sillence, como osteogénesis imperfecta tipo 2-A y 2-B, respectivamente). Muslos mantenidos en abducción fija y rotación externa con limitación del movimiento de la mayoría de las articulaciones. Disminución de la osificación del cráneo, fracturas múltiples de huesos largos y costillas. Tórax pequeño. Vértebras hipoplásicas y trituradas10.
La muerte en la osteogénesis imperfecta tipo II ocurre por fallo respiratorio durante las primeras horas de vida. Más de 60% de los recién nacidos mueren en el primer día de vida, el 80% restante muere durante el primer mes, y la supervivencia más allá del año es rara. El patrón respiratorio restrictivo se relaciona con la severidad de la enfermedad.
El diagnóstico prenatal es posible a partir de las 17 semanas por medio de ecografía. También se puede analizar la síntesis de procolágeno en las células del líquido amniótico. Si se conoce la mutación, se puede realizar el estudio genético por biología molecular de vellosidades coriales o líquido amniótico.
El análisis de la mutación (y la importancia de determinarla) es la mejor forma de diagnóstico prenatal por lo temprano y certero del diagnóstico, además permite un diagnóstico genético preimplantacional.
El diagnóstico prenatal de esta paciente se basó en las ecografías obstétricas morfoestructurales que informaban una probable displasia esquelética.
Nuestra paciente presenta una forma de osteogénesis imperfecta neonatal severa o tipo II por sus manifestaciones clínicas y las características radiográficas. Diagnóstico que se apoya en la autopsia clínica y estudio genético.
En la actualidad no existe tratamiento curativo. El objetivo del tratamiento en los pacientes con osteogénesis imperfecta es reducir la incidencia de fracturas, minimizar el dolor y maximizar la movilidad. La fisioterapia, la rehabilitación y la cirugía son complementos adicionales al tratamiento9,12,13. Los pacientes con osteogénesis imperfecta tipos I y IV generalmente solo requieren atención ortopédica de rutina para sus fracturas y asesoramiento sobre prevención de accidentes y seguridad. Los cuidados paliativos de apoyo y el manejo mínimo son las únicas medidas disponibles para los bebés con osteogénesis imperfecta tipo II. El tratamiento de bebés y niños con osteogénesis imperfecta tipo III está orientado a minimizar la frecuencia de fracturas y prevenir deformidades. En la infancia, esto puede significar un manejo limitado del niño y el uso de un dispositivo de transporte acolchado. Posteriormente, puede ser necesario tratamiento quirúrgico en forma de osteotomías y estabilización interna de los huesos largos. El mantenimiento de la actividad y evitar períodos prolongados de inmovilización ayudan a prevenir la atrofia por desuso. El papel del tratamiento con bifosfonatos en el cambio de la historia natural de la osteogénesis imperfecta no se comprende completamente. El tratamiento con bifosfonatos no pareció reducir la incidencia de fracturas, pero sí afectó la densidad ósea y la altura de los adultos. Además, la terapia con hormona del crecimiento humano como complemento de la terapia con bifosfonatos se correlacionó con un mejor crecimiento lineal y un aumento de la densidad mineral ósea. La hormona de crecimiento, que estimula el metabolismo de los huesos y el colágeno, ha mostrado cierta promesa en niños con formas más graves de osteogénesis imperfecta tipo I. Sin embargo, está contraindicada en pacientes con cifoescoliosis porque aumenta la tasa de progresión de la deformidad y también está contraindicada en niños con OI tipos II a IV debido a su efecto estimulante sobre el metabolismo del colágeno, solo sirve para aumentar la secreción de colágeno anormal3,5,14-16.
Cuando se sospecha el diagnóstico prenatal de osteogénesis imperfecta severa, se deben tomar todas las medidas preventivas para minimizar las posibles complicaciones. La vía de nacimiento de elección según consenso es la operación cesárea; sin embargo, esto no aumenta la supervivencia y no altera la tasa de fracturas5,11,17.
El tratamiento en esta paciente se basó en medidas de confort, con sedoanalgesia adecuada según escalas del dolor aprobadas y apoyo ventilatorio con asistencia ventilatoria mecánica desde el nacimiento.
Conclusiones
La osteogénesis imperfecta tipo II es la forma más grave de presentación, con una alta mortalidad en el período perinatal. La sospecha diagnóstica prenatal es posible, con ecografías morfoestructurales que observen acortamiento de huesos largos, remodelación de éstos y pobre mineralización. La confirmación luego del nacimiento es en base a las características clínico-radiológicas (evidenciando las múltiples fracturas en distintos tiempos evolutivos); en el presente caso clínico, el estudio genético y necrópsico apoyan el diagnóstico de osteogénesis imperfecta tipo II.
No existe actualmente tratamiento curativo, las medidas implementadas son sostén ventilatorio, confort y sedoanalgesia.
La consejería genética y planificación de próximos embarazos se vuelve muy importante en esta familia, dado el antecedente de cinco abortos espontáneos previos. El estudio genético de los progenitores es aconsejable para descartar mosaicismo gonadal y determinar si la variante fue heredada o aparece en la paciente como un suceso de novo. En base a estos resultados, valorar el riesgo o probabilidad de recurrencia.

