











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
PermalinkIntroducción
Las epidermólisis bullosas (EB) corresponden a un grupo de genodermatosis infrecuentes clínica y genéticamente heterogéneas, caracterizadas por fragilidad cutánea leve a severa y ampollas o erosiones, luego de traumas mínimos1. Defectos genéticos que alteran moléculas esenciales de las uniones epidérmicas conducen a una pérdida de la capacidad de adhesión de la membrana basal2. La gravedad de las manifestaciones clínicas varía mucho entre los tipos y subtipos de EB, abarcando un espectro que va desde ampollas pequeñas hasta erosiones extensas, secuelas cicatrizales y complicaciones mortales.
La severidad de la presentación clínica en la etapa neonatal no se correlaciona con el pronóstico. Es una enfermedad crónica con gran impacto en la calidad de vida que requiere un abordaje multidisciplinario.
La incidencia reportada varía por zona geográfica, con una incidencia mundial estimada de uno cada 20.000 habitantes3).
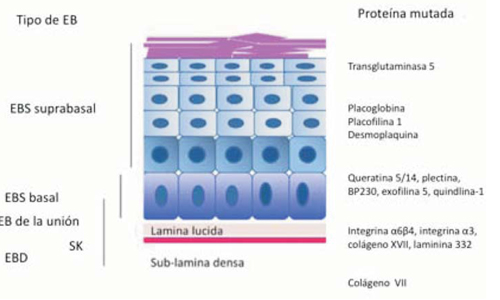
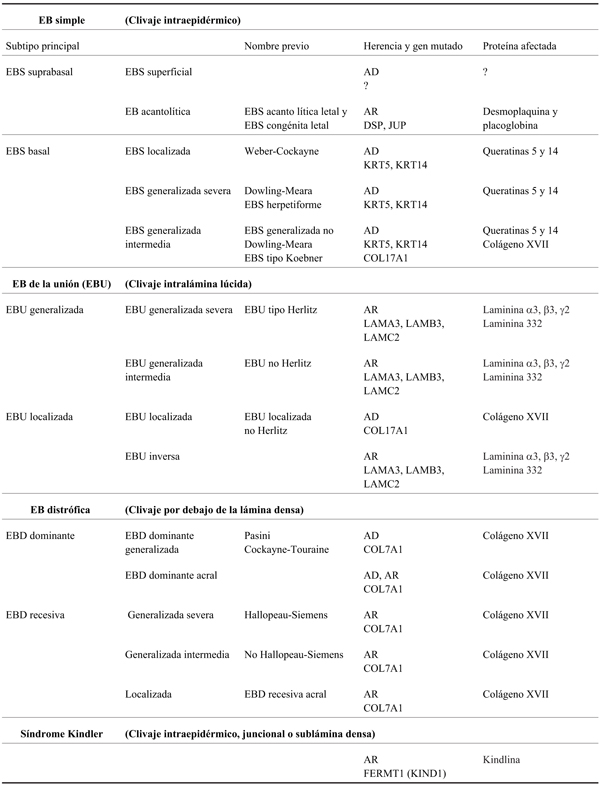
Se describen cuatro tipos mayores de EB: simple (EBS), de la unión (EBU), distrófica (EBD) y síndrome Kindler4. Se diferencian entre sí de acuerdo al sitio de separación y formación de las ampollas en la piel, ya sea suprabasal, en la zona de la membrana basal o infrabasal5 (Figura 1). Cada tipo abarca varios subtipos según extensión (localizada o generalizada) y según qué molécula de la piel se encuentra afectada5. Esta clasificación surge de reuniones de consenso, la última en 2013, que agrega una subclasificación enfocada en el origen molecular de cada subtipo4 (Tabla 1). Existen nuevas y más complejas clasificaciones, como el abordaje “en piel de cebolla”, que considera secuencialmente el tipo de EB, el modo de herencia, fenotipo, mapeo antigénico por inmunofluorescencia (IF) y mutaciones presentes en cada paciente4.
Es indispensable un correcto manejo de estos pacientes desde el nacimiento, así como la asistencia y contención durante la vida. Con este propósito existe una organización internacional, la fundación DEBRA (Dystrophic Epidermolisis Bullosa Research Association), una asociación sin fines de lucro, dedicada al apoyo de los enfermos con “piel de cristal” y su familia. Promueve la investigación y la difusión del conocimiento de la enfermedad6. Basados en las pautas de manejo de esta organización se han creado pautas nacionales ajustadas a nuestro medio.
Actualmente no hay terapia efectiva o cura para la EB4. El tratamiento es sintomático, aunque se están investigando nuevas terapias celulares y moleculares7.
Caso clínico
Presentamos el caso de un neonato de 15 días de vida de sexo femenino, procedente del departamento de Artigas, producto de segunda gesta.
Embarazo bien controlado y tolerado. Sin complicaciones. Parto vaginal, recién nacido de término, vigoroso, con un peso adecuado para la edad gestacional. Calmetizado. Alimentado con pecho directo exclusivo.
Presenta antecedente familiar (AF) de padre con EBS con buena evolución.
La paciente fue derivada a la unidad de Dermatología Pediátrica del Centro Hospitalario Pereira Rossell (CHPR) por una dermatosis que se inicia a los tres días de vida, con lesiones que aparecen frente a la fricción y a traumatismos mínimos. Al examen físico presenta una dermatosis diseminada, topografiada en cabeza y miembros, con predominio acral, localizada en dedos de manos y pies, y en ambos talones. Caracterizada por ampollas tensas y vesículas con contenido claro que asientan sobre piel sana y lesiones con costra serohemática (Figura 2). A nivel de las mucosas hay una pequeña erosión a nivel de paladar duro, sin otras lesiones. A nivel de las faneras, onicodistrofia de algunas uñas de los pies. Paciente con buen estado general y en apirexia. Sin dismorfias. Dado el AF de EBS y la clínica de la paciente y con sospecha de EBC, se realiza una biopsia de piel para microscopía electrónica que informa clivaje a nivel del estrato basal con clumps de tonofilamentos compatible con EBS tipo Dowling-Meara, según la clasificación de la DEBRA 2008. Valorado por Gastroenterología, no evidencia lesiones en paladar blando, buena succión, tránsito digestivo normal. Se indican medidas higiénicas basadas en el cuidado de la piel evitando traumatismos, baños con limpiadores syndet, luego del baño aplicar xtericol sobre lesiones con gasas vaselinadas y sobre ellas red elástica, relevo bacteriológico de lesiones cada 15 días, pecho directo exclusivo, se explican síntomas y signos de reconsulta precoz y se realiza interconsulta con genetista.
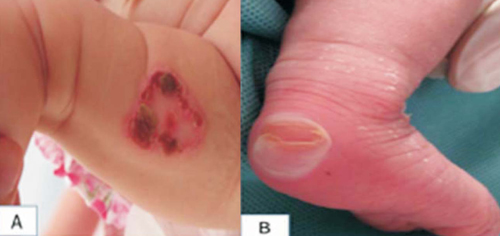
Figura 2: A) Collarete epidérmico con costra serohemática a nivel de cara interna de muslo. B) Ampolla epidérmica semidestechada a nivel de talón.
Buena evolución de las lesiones cutáneas con el tratamiento instaurado, otorgándose el alta con controles periódicos. Se destaca que ha presentado buen crecimiento y desarrollo, y con respecto a las lesiones cutáneo-mucosas, destacamos que hasta el momento no ha presentado nuevas lesiones en mucosa oral, y con referencia a la esfera cutánea ha evolucionado con empujes y remisiones a nivel predominantemente acral, sin secuelas cicatrizales, y con un buen manejo por parte de los padres y apoyo del equipo multidisciplinario médico, sin mayor repercusión en su calidad de vida.
Discusión
El diagnóstico de EBC se sospecha en niños con piel frágil y se manifiesta por la formación de ampollas o erosiones ante traumatismos menores. Es importante una correcta anamnesis y examen físico para sospechar el diagnóstico5. Debe diferenciarse de otras múltiples enfermedades que se presentan con ampollas, principalmente descartar lo infeccioso, luego otras (dermatitis herpetiforme, dishidrosis, ampollas por fricción, etcétera). Para confirmar el diagnóstico se realiza una biopsia de piel de una lesión reciente para su estudio anatomopatológico y con IF o ME, que permitirá determinar el plano de formación de la ampolla dentro de la piel5. El mapeo por IF permite determinar el plano de despegamiento e identificar la proteína afectada. La ME es el método de referencia en la clasificación de la EB, pudiendo determinar el plano de despegamiento y las alteraciones ultraestructurales de las proteínas afectadas7. Puede realizarse el estudio genético del paciente y de sus padres para determinar el tipo de mutación y de herencia que permitirán realizar consejería genética y establecer el pronóstico5.
Clasificación
Con enfoque práctico describiremos el tipo de EB de nuestra paciente y brevemente las otras formas.
Epidermólisis bullosa simple
Es la forma de presentación más frecuente (92%)5,8. La mayoría es de herencia autosómica dominante y se debe a mutaciones en las queratinas 5 o 14, o ambas4,5,9. La formación de ampollas ocurre con mayor frecuencia en la infancia y disminuye con la edad, no suelen dejar cicatriz5.
Existen múltiples variantes, siendo la más frecuente la EBS localizada (antes denominada de Weber-Cockayne). Suele comprometer manos y pies, se desarrolla en la infancia temprana con el inicio de la deambulación, aunque en algunos casos puede manifestarse más tardíamente en la adolescencia o en el adulto joven4,9,10).
Se manifiesta por ampollas con base eritematosa originadas por fricción y exacerbadas por la sudoración y el calor excesivos9-12. Otra variante es la denominada EBS generalizada intermedia (antes denominada de Koebner). Puede presentarse en el nacimiento o en los primeros meses de vida, con ampollas y erosiones en sitios de roce, las cuales curan sin dejar cicatriz, puede acompañarse de lesiones mucosas orales4,9,13. El compromiso es más diseminado que la forma localizada, pero menos severa que la EBS generalizada severa13. La EBS generalizada severa, antes conocida como de Dowling Meara, presenta gran variabilidad clínica entre los individuos afectados4,9. Presente desde las primeras semanas de vida, se caracteriza por ampollas herpetiformes con base eritematosa2,9. Las manos y los pies son los sitios de predilección, si bien las lesiones pueden ser diseminadas. Característicamente las lesiones de palmas y plantas son seguidas por queratodermia focal. Suele acompañarse de engrosamiento de uñas (paquioniquia) y el compromiso mucoso oral no suele ser severo. Las ampollas pueden ser excepcionalmente severas en el período neonatal, pudiendo ocasionar la muerte, principalmente por sepsis9. Otra variante severa es la EAS letal acantolítica, según la última clasificación descrita como EAS acantolítica, debida a la mutación autosómica recesiva del gen de la desmoplaquina, proteína de adhesión en las células epiteliales y musculares. La afección es generalizada y se manifiesta en el posparto inmediato con fragilidad cutánea, alopecia universal, onicomadesis y presencia de dientes neonatales. No aparecen ampollas o vesículas, predominando las erosiones. Se constatan erosiones mucosas conjuntivales y en cavidad oral así como alteraciones genitourinarias, pudiendo presentar también alteraciones gastrointestinales12. En casos raros la EAS puede acompañarse de distrofia muscular o atresia pilórica, o pigmentación moteada configurando otros subtipos12,14.
Epidermólisis bullosa de la unión
La separación de la piel ocurre en la unión dermoepidérmica a nivel de la membrana basal, en la lámina lúcida15 (Figura 1). Es la forma menos frecuente de EB. Caracterizada por presentar erosiones y ampollas generalizadas a nivel cutáneo y mucoso, distrofia ungueal, hipoplasia del esmalte dental y caries16. Las lesiones dejan cicatrices atróficas así como quistes de milio17. Las formas con compromiso mucoso pueden acompañarse de anemia, retraso del crecimiento y alteraciones respiratorias. La principal causa de muerte en estos pacientes es por sepsis seguida de fallo respiratorio18. La forma más común y grave de EBU es la generalizada severa. Estos pacientes no suelen sobrevivir a la infancia19.
Epidermólisis bullosa distrófica
Se debe a mutaciones en el colágeno VII que forma las fibrillas de anclaje de la membrana basal epidérmica (Figura 1). Se describen tres variantes con severidad creciente: EBD generalizada dominante, EBD recesiva intermedia y EBD recesiva severa1. Los síntomas cardinales de la mayoría de las formas dominantes o recesivas son fragilidad cutánea, ampollas, cicatrices, quistes de milio y alteraciones ungueales. Las formas generalizadas severas llevan a contracturas articulares, pseudosindactilia, mutilaciones, compromiso mucoso extracutáneo, malnutrición y retraso del crecimiento. En estas también ocurre la mayor incidencia de carcinoma espinocelular, con inicio temprano (desde segunda década) y comportamiento agresivo1.
Síndrome de Kindler
Se debe a la mutación del gen KIND1 que codifica para una proteína, kindlina, cuya función es estabilizar la unión dermoepidérmica. Se manifiesta en el período neonatal y en la infancia por la formación de ampollas acrales y fotosensibilidad, con poiquilodermia en años posteriores1,5). La formación de ampollas puede ser espontánea o traumática y en general se resuelve con la edad. Puede existir atrofia cutánea generalizada, siendo más común en manos, pies, rodillas y codos17.
Tratamiento
Requiere un abordaje multidisciplinario enfocado en el manejo de las heridas; dolor, prurito, limitar las complicaciones infecciosas cutáneas, cicatrices y cáncer de piel; mantener movilidad y estado nutricional adecuados20,21. Es importante el correcto manejo desde el momento de la recepción de estos neonatos para mejorar la salud y calidad de vida, brindando lineamientos a familiares y cuidadores, minimizando dudas e incertidumbres que se presenten en la vida cotidiana.
El manejo de heridas requiere valoración de la extensión de piel comprometida, morfología (ampollas intactas versus erosiones) y la presencia de exudado. Durante el cambio de curaciones, estas se deben remojar con solución fisiológica previo a su remoción y aplicar emoliencia en forma previa a la colocación de la nueva curación. Cuando hay ampollas, es fundamental puncionarlas con aguja estéril para reducir la extensión de la separación dermoepidérmica, manteniendo el techo remanente, ya que este provee de una protección natural21. En erosiones no exudativas se pueden utilizar espumas no adherentes o parches de hidrocoloide. En heridas mínimamente exudativas se recomienda el uso de hidrogeles (Tegaderm®), que ayudan al alivio del dolor y prurito. Para heridas exudativas se deben utilizar hidrofibratos o alginato de calcio (Kaltostat®) y en heridas colonizadas o infectadas antimicrobianos, como apósitos embebidos en sulfadiazina de plata (Actisorb®) en caso de niños mayores. Son frecuentes en la EB heridas con evidencia de colonización bacteriana, requiriendo el uso repetido de antibióticos tópicos o sistémicos, estos deberán ser usados solo en casos de infección, evitando su uso profiláctico para evitar resistencias bacterianas21. Hasta un 90% de las heridas crónicas de EB están colonizadas por S. aureus22. Se puede agregar al agua del baño sal e hipoclorito de sodio con el objetivo de prevenir la infección. Se ha demostrado que los baños en agua salada reducen el dolor (91%), el uso de analgésicos (66%), la fetidez (31%) y el exudado (44%) en todos los tipos de EB23.
Es importante el adecuado manejo del dolor en estos pacientes. Para su tratamiento se recomiendan antiinflamatorios no esteroideos (AINES) y acetaminofén, y cuando la severidad lo indica se puede utilizar tramadol y opiodes21,24. Para el dolor neuropático, a veces descrito por los pacientes, se utiliza gabapentina con buenos resultados21,24,25. Estos pueden combinarse con medidas no farmacológicas, como psicoterapia o técnicas de relajación, entre otras21,24.
El prurito predomina en la noche interfiriendo con el sueño26,27. Para el tratamiento se utilizan antihistamínicos sedantes y no sedantes, antidepresivos, gabapentina y pregabalina, asociados a una buena emoliencia21.
En los casos con cicatrices y contracturas articulares que generen restricción de movimientos y limitación funcional es indispensable la fisioterapia, pudiendo algunos pacientes llegar a requerir corrección quirúrgica21).
La prevención de heridas cutáneas es importante en vistas al desarrollo motor e integración social de los niños. Por ejemplo, pueden utilizarse almohadillas de neopreno para proteger extremidades cuando aprenden a gatear, o colocación de telas acolchadas en asientos de triciclos y otros juegos28).
Las heridas crónicas y sus complicaciones, como la pérdida insensible de agua, inestabilidad térmica e infecciones, llevan a un estado inflamatorio hipercatabólico, por lo que es necesario aumentar el aporte calórico29. El déficit nutricional se correlaciona con la severidad de la EB, siendo entonces más severo en EBD que en EBU.
Es importante la monitorización de cicatrices crónicas o heridas que no curan en EBD y EBU por el riesgo aumentado de melanoma y cáncer de piel no melanoma. Se deben biopsiar las lesiones sospechosas. El screening cada 6-12 meses debe comenzar a partir de los 20 años. Instruir para autoexamen de piel21).
Es necesaria una aproximación terapéutica curativa, actualmente en fase experimental. Algunos procedimientos en estudio incluyen terapia génica, proteica y celular, y el cultivo y trasplante de queratinocitos30-32).
Conclusión
La EB es una enfermedad compleja que requiere de un cuidado adecuado desde el nacimiento y un manejo multidisciplinario para mejorar calidad de vida y reducir la morbimortalidad de estos pacientes. El diagnóstico es clínico presuntivo de confirmación histopatológica con IF o ME, apoyado por el AF. Actualmente, contamos con pautas nacionales del manejo del recién nacido con piel frágil, las cuales han sido desarrolladas con la finalidad de ayudar al personal de salud en el manejo y cuidado de estos niños31. Es importante el adecuado cuidado de las heridas, control del dolor y prurito desde el momento del nacimiento, así como prevenir potenciales complicaciones de la enfermedad. Las terapias moleculares actualmente en estudio podrían en un futuro modificar el pronóstico de estos pacientes.

