











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
PermalinkIntroducción
La linfohistiocitosis hemofagocítica (LH) se describió por primera vez en 1939, como una condición caracterizada por fiebre, adenopatías, pancitopenia y hemofagocitos en medula ósea1.
En 1952 se vincula esta entidad a un desorden inmunitario familiar, denominándose reticulocitosis familiar hemofagocítica. Sin embargo, si bien su origen puede estar asociado a herencia autosómica recesiva, se describieron posteriormente formas secundarias, asociadas a infecciones, enfermedades malignas, fármacos o enfermedades autoinmes1.
Este síndrome se caracteriza por una respuesta inmune desproporcionada e inefectiva consecuencia de la disfunción de las células Natural killer (NK) que conduce a sobreestimulación, proliferación y migración ectópica de linfocitos T2. la fisiopatología aún tiene aspectos que no están del todo claros, sin embargo en los casos familiares esta alteración en la regulación del sistema inmune se relaciona con defectos en la actividad citotóxica mediada por una proteína llamada perforina de las células T CD8 y NK que determinan una respuesta inflamatoria que se perpetúa, provocando las manifestaciones clínicas de esta enfermedad que inicialmente son similares a otras de mayor prevalencia, dificultando y retrasando el diagnóstico3,4.
La incidencia se estima en 1-2 casos/millón de individuos/ año, planteándose que existe un subdiagnóstico. Los casos familiares se presentan en 1:50.000 recién nacidos vivos y la sobrevida es inferior a dos meses sin mediar tratamiento, siendo una infección el desencadenante de la misma5.
En 1994, la Sociedad de Histiocitosis publica el primer protocolo internacional de tratamiento y en 2004 se presentaron las modificaciones con la información analizada durante diez años (1994-2004), incluyéndose los nuevos criterios diagnósticos y guías terapéuticas2.
El objetivo de la presentación de este caso clínico es concientizar a los pediatras de la existencia de esta entidad, para pensar en ella en determinadas situaciones clínicas que así lo justifiquen, llegando a un diagnóstico precoz que permita iniciar el tratamiento oportuno, efectivo y dirigido.
Caso clínico
Varón, 4 años, previamente sano. Consulta en emergencia por enfermedad de seis días evolución con fiebre de hasta 37,8 ºC y decaimiento. Agrega dolor abdominal difuso, rechazo del alimento, dolor miccional, orinas anormalmente coloreadas, edema de párpados, dolor en miembros inferiores que le impiden la deambulación, fatiga, tos y mal estado general. Al examen reactivo, ventilando al aire (VEA) satura 95%, polipneico, edema de cara, manos y pies fríos, blancos, indoloros. Hipoventilación y matidez basal derecha. Abdomen depresible, indoloro, sin visceromegalias. Apirético.
Se realizó radiografía de tórax (Figura 1) que informó opacidad homogénea de base de hemitórax derecho, con aumento de los espacios, con curva ascendente, compatible con derrame pleural y cardiomegalia.
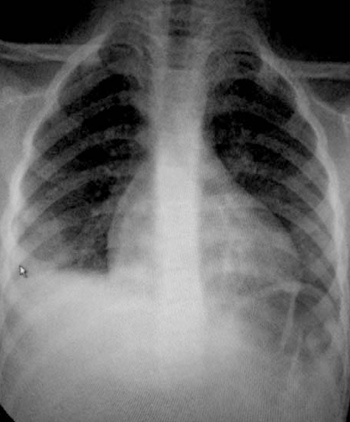
De la analítica: anemia (Hb 7,2 g/dl, Ht 21%), leucopenia (2.100 elementos/mm3 con 67% neutrófilos), plaquetopenia (22.000 elementos/mm3). La lámina periférica objetivó desviación a izquierda de serie blanca, escasas plaquetas. No blastos. PCR elevada: 242 mg/dL. Alteración de funcional hepático con aumento de LDH (13.500 U/L), bilirrubina directa (3.80 mg/dl) fosfatasa alcalina (1036 U/L), TGP (127 U/L), TGO (64 U/L), GGT (205 U/L). Hipoalbuminemia (2,04 g/dl). Crasis alterada con hipoprotrombinemia (53%) e hipofibrinogenemia (53 mg/l). Hiponatremia (128 mEq/l).
Diagnóstico inicial: sepsis a punto de partida respiratorio. Neumonía de inicio agudo, comunitaria grave con derrame pleural. Se inicia tratamiento antibiótico y se traslada a cuidados intensivos (UCIP).
Al ingreso: GSC 15, VEO2 por CN 3 l/m, SatO2 92%, polipneico, quejido espiratorio, a nivel pleuropulmonar asimetría torácica ya referida. Hemodinamia estable. Ictericia. Petequias en territorio axilar, zonas de punción y algunas difusas. Hepatopesplenomegalia.
Se completa valoración infecciosa con hemocultivos, marcadores virales para hepatitis A, B y C, CMV, parvovirus B19, herpes virus e hisopado nasofaríngeo que serán negativos. Toracocentesis diagnóstica y terapéutica bajo ecografía y luego de concentrado plaquetoria evidencia líquido con las características de un trasudado cuyo cultivo será negativo para bacterias, hongos y parásitos.
El mielograma evidenció la presencia de un fenómeno hemofagocítico y se completó valoración con triglicéridos (250 mg/dl) y ferritina (100.000 μg/ml), ambos elevados. El tratamiento inicial fue con antibióticos, inmunoglobulinas, metilprednisolona y estimulantes de colonias granulocíticas, agregándose a las 24 horas con el resultado del mielograma etopósido y ciclosporina según el protocolo HLH 2004 de la Sociedad de Histiocitosis.
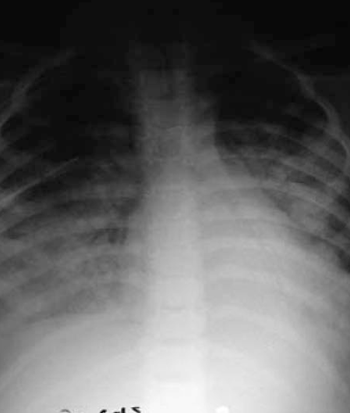
Se inicia oxigenoterapia de alto flujo (OAF) con rápido escalamiento a ventilación invasiva por gravedad clínica con compromiso del estatus neurológico y respiratorio, evolucionando al síndrome de dificultad respiratoria (SDRA) (Figura 2).
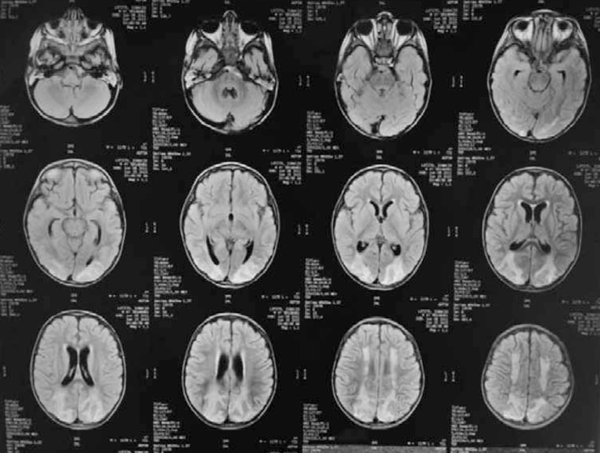
Ecocardiograma con Doppler: fallo contráctil de ventrículo izquierdo que mejora con diuréticos e inotrópicos. RNM de cráneo (Figura 3) evidencia extensas lesiones cortico-subcorticales bilaterales en centro oval y fronto-parietales homogéneas con áreas hipo e hiperintensas en T1, T2 y Flair, con espacios de restricción de difusión y realce gírico con el contraste. Ventrículos de amplitud ligeramente aumentada, al igual que los espacios subaracnoideos.
Permaneció internado 48 días, mejorando progresivamente las alteraciones clínicas y paraclínicas, destacándose la afectación neurológica con hipertonía de los cuatro miembros, hiperreflexia, clonus agotable al estímulo y hemiparesia de hemicuerpo derecho. Luego de logrado el destete de la ventilación, se mantuvo vigil, sin ningún vínculo con el ambiente, en posición fetal, planteándose además de la encefalopatía vinculada a la LH la posibilidad de un síndrome de encarcelamiento o posinternación en CTI iniciándose risperidona con buena respuesta cognitiva, manteniendo el compromiso motor sobre todo de miembros inferiores. Potenciales evocados auditivos y visuales normales.
Discusión
La LH engloba un grupo heterogéneo de enfermedades que pueden manifestarse a cualquier edad. La forma genética (primaria) usualmente se manifiesta antes del primer año de vida, en 70%-80%, mientras que la forma adquirida o secundaria está relacionada predominantemente con procesos infecciosos, oncológicos, reumatológicos, inmunológicos o fármacos (Tabla 1)5.
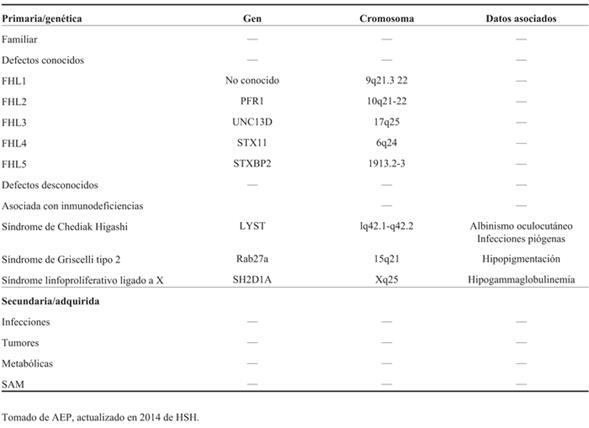
Se relaciona con una inmunorregulación anormal, la que contribuye a una respuesta inmune exagerada y descontrolada. En las formas genéticas la transmisión es autosómica recesiva. En las secundarias se identifican neoplasias (linfomas, leucemias, síndromes mielodisplásicos, carcinomas y tumores de células germinales), infecciones por gérmenes con replicación intracelular (virus: Epstein-Barr, citomegalovirus, varicela-zóster, herpes simple, sarampión, arbovirus, herpes 6, adenovirus, parvovirus y dengue), bacterianas sistémicas, incluso tuberculosis miliar, brucelosis y tifus, Haemophylus, Serratia y Legionella. En cuanto a las infecciones fúngicas, candida e histoplasma son las más representativas, y de las parasitarias destacamos leishmaniasis y toxoplasmosis6. En muchos casos se demostró una inmunodeficiencia subyacente, proponiéndose que sería un déficit en la inmunidad celular específica la que determinaría, ante un estímulo adecuado antigénico, la proliferación y activación histiocítica de una manera compensadora, lo que no sucedió en este caso.
El diagnóstico puede realizarse por la detección de mutaciones genéticas o por la presencia de cinco de los ocho criterios diagnósticos aceptados, propuestos por la Histiocyte Society (Tabla 2)7.

La fiebre y la esplenomegalia están presentes en el 75% de los casos al momento del diagnóstico; la bicitopenia, hipertrigliceridemia y ferritina >500 ng/mL 50% y la hemofagocitosis entre 25%-100%2, como sucedió en este caso.
La presentación clínica es variable, siendo fundamental el alto índice de sospecha y el seguimiento de estos niños8.
Las manifestaciones dérmicas son variadas, en este caso la presencia de petequias o un exantema maculopapular que puede verse entre 6%-65% de los casos. El desarrollo de fallo pulmonar, también presente, empeora el pronóstico y sugiere un inadecuado control del síndrome o del proceso infeccioso cuando está la causa desencadenante.
En una serie publicada de 11 pacientes entre 5 meses y 13 años, con LH y fallo multiorgánico (FOMS)9, el compromiso respiratorio estaba presente en siete pacientes con evolución al SDRA, como sucedió en este niño. El cultivo de secreciones negativo, junto a histiocitos activados con fagocitosis pulmonar en la biopsia y el líquido pleural (no evidente en este paciente), apoyan el compromiso inicial pulmonar. En este mismo trabajo se observó que la afectación cardiovascular estuvo presente en cinco pacientes, manifestándose como shock que requirió elevadas dosis de inotrópicos y vasopresores. El fallo contráctil estaba presente en el caso que presentamos con rápida mejoría. El FOMS para estos autores está vinculado a niveles elevados de citoquinas junto con la infiltración por histiocitos activados de diversos tejidos. La tercera parte de la LH tiene manifestaciones neurológicas, como pasó en este paciente con deterioro del nivel de conciencia y ataxia. En un 50% el líquido cefalorraquídeo (LCR) será patológico y la neuropatía periférica difusa secundaria a la destrucción de mielina por los macrófagos activados está presente en el 70% de los casos10. La patogénesis es desconocida, pero se vincula a un aumento de secreción de citoquinas Th1, interferón gamma e Il-2, activándose las células T y la línea de monocitos/macrófagos (Figura 4).
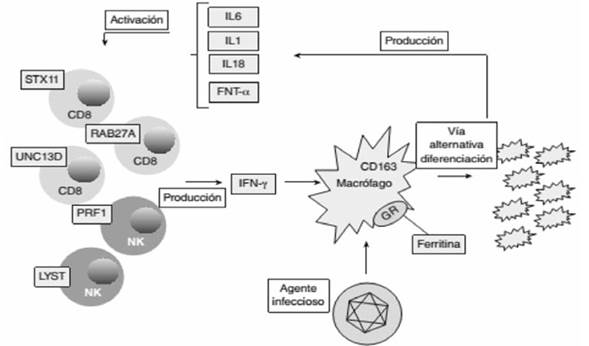
Figura 4: Fisiopatología del síndrome hemofagocítico que se caracteriza por una respuesta inmune desproporcionada. Tomado de Espinosa y colaboradores.
El diagnóstico oportuno favorece el inicio de un tratamiento específico que mejorará la mortalidad, tan elevada en esta entidad, que incluye inmunosupresores, agentes moduladores, anticuerpos monoclonales, el tratamiento de la enfermedad desencadenante y en algunos casos trasplante de progenitores hematopoyéticos, dependiendo el mismo de que sea una forma familiar o secundaria a infección, enfermedad maligna o de etiología autoinmune. El mismo se basa en el protocolo HLH-2004, modificado por la Histiocytic Society que incluye dos etapas:
- Terapia de inducción, que incluye el uso de dexametasona, etopósido y ciclosporina con o sin quimioterapia intratecal, la que se sugiere cuando el compromiso neurológico es grave, como sucedió en este paciente. Es fundamental iniciarlo precozmente si las infecciones o citopenias no han sido resueltas. Si responden en forma adecuada el protocolo se suspenderá luego de un período mantenido de remisión y en caso de reactivación se reiniciarán etopósido y dexametasona, incrementándose en forma progresiva hasta obtener nuevamente la respuesta.
- Terapia de rescate, se iniciará en aquellos pacientes que no presentan ni siquiera una respuesta parcial (mejoría de los parámetros de laboratorio en un 25%) luego de 2-3 semanas de comenzada la inducción. El fármaco recomendado teniendo en cuenta el papel que juegan las células T en la etiopatogenia de esta enfermedad son los anticuerpos monoclonales como el alembuzumab.
La respuesta completa se logra en 50% de los casos; la respuesta parcial en 30% y la muerte se producirá en 20%.
En la etapa de inducción en este paciente se utilizaron inicialmente pulsos de metilprednisolona a 30 mg/kg/día por tres días continuando con dexametasona. Ante la no mejoría se agrega etopósido, ciclosporina e inmunoglobulinas. La recomendación del etopósido es en los pacientes de alto riesgo, como son los que presentan afectación del SNC, insuficiencia renal severa, FOMS o que no tuvieron mejoría con la metilprednisolona como sucedió en este caso. La eficacia de la inmunoglobulina es discutida, pero en los casos graves recomiendan su utilización11.
Finalizado el tratamiento en este caso y por la respuesta obtenida se mantuvo el mismo manejo. Sin embargo, en algunos pacientes con respuesta parcial se plantea el trasplante alogénico de progenitores hematopoyéticos12.
Entre los factores de mal pronóstico en la LH están el retraso diagnóstico, el fallo orgánico múltiple, la presencia de coagulopatías y/o neutropenia severa, afectación del SNC y mala respuesta al tratamiento. En este niño estaban todos presentes13. Pese a los cambios realizados en el tratamiento desde 1990, el compromiso neurológico aumenta la mortalidad hasta en el 20% de los casos y de los que sobreviven hasta un 60% quedarán con secuelas neurológicas13.
En el momento actual y luego de dos años del alta, el desarrollo cognitivo es adecuado para su edad, mantiene compromiso motor de miembros inferiores que ha mejorado progresivamente con fisioterapia y seguimiento con pediatra y hematooncólogo.
Conclusiones
La LH no es frecuente en pediatría, pero teniendo en cuenta la relevancia del diagnóstico y del tratamiento oportunos debe ser sospechado por la clínica, con la presencia de marcadores inflamatorios elevados como ferritina, PCR, fiebre persistente y esplenomegalia. El tratamiento debe incluir el de la causa desencadenante cuando es clara, los fármacos para bloquear la respuesta inflamatoria sistémica y el de sostén de los sistemas y órganos en falla.

