











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
PermalinkIntroducción
Las anomalías congénitas de las arterias coronarias son raras en la población general, con una incidencia de solo 0,3%-0,9% en necropsias. Se observan hasta en 5% de pacientes sometidos a angiografía coronaria. pudiendo incrementarse a 36% en aquellos con enfermedades congénitas cardíacas. Los reportes en la edad pediátrica son pocos1-3. Si bien su espectro de presentación clínica es amplio, desde ser asintomática a una presentación letal, es necesario su reconocimiento en ciertas circunstancias al ser una de las principales causas de muerte durante el ejercicio en jóvenes y una complicación a tener en cuenta en la reparación de cardiopatías congénitas4. Es responsable de un tercio de las muertes por enfermedad coronaria no ateroesclerótica5. El origen anómalo de la arteria coronaria derecha desde el tronco de la arteria pulmonar (Anomalous origin of the right coronary artery originating from the pulmonary trunk, ARCAPA); descritos los primeros casos por Brooks en 18856,7, es extremadamente rara con una estimación en la población general de 0,002% y al ser muchos pacientes asintomáticos su prevalencia real debe ser seguramente subestimada. El diagnóstico es habitualmente accidental, como en nuestro caso, o basándose en la existencia de un soplo cardíaco, dolor torácico, arritmias, fallo cardíaco congestivo e infarto miocárdico. Solamente 25% a 30% de los casos se asocian a defectos cardíacos congénitos, siendo la tetralogía de Fallot, la ventana aortopulmonar, la estenosis aórtica y los defectos septales las asociaciones más frecuentemente encontradas.
La ecografía transtorácica es la exploración no invasiva más difundida para su detección. La terapia definitiva requiere del reimplante de la arteria desde su origen anómalo a la raíz aórtica2,6.
Observación clínica
J.R. Sexo femenino. Seis meses. Madre de 25 años, sin antecedentes. Producto de primera gestación. Embarazo gemelar, monocorial, monoamniótico. Bien controlado y tolerado hasta las 33 semanas, donde se evidencia Doppler alterado en ambos fetos. Cesárea. Segunda gemelar, pretérmino de 33 semanas. Vigorosa. Peso al nacer 1.726 g. Síndrome de dificultad respiratoria adaptativo. Evolución neonatal sin complicaciones. Ecocardiograma de control evidencia probable origen anómalo de arteria coronaria derecha desde tronco de arteria pulmonar (Figura 1). Cateterismo cardíaco diagnóstico realizado a los 4 meses de vida muestra una arteria coronaria derecha dilatada que nace desde el tronco de arteria pulmonar en su cara anterior, circulación intercoronaria importante. Arteria coronaria izquierda de nacimiento normal.
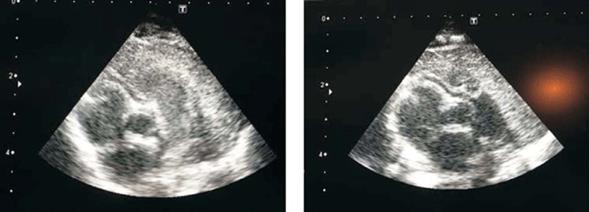
Figura 1: Ecografía transtorácica realizada en el período neonatal que evidencia un probable origen de la arteria coronaria derecha emergiendo desde el tronco de la arteria pulmonar con flujo desde esta a la coronaria.
A los seis meses se realiza reimplante de arteria coronaria derecha en posición de sigmoidea derecha. Plastia de tronco de arteria pulmonar con parche de pericardio autólogo. Hemodinamia estable en posoperatorio. Asistencia ventilatoria por 24 horas. Egreso a domicilio sin medicación cardiológica.
Discusión
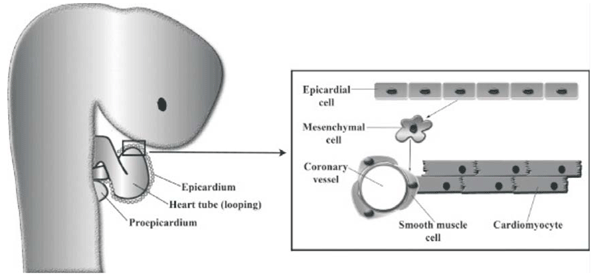
El origen y los mecanismos de desarrollo de los vasos coronarios no son del todo comprendidos8. El sistema arterial coronario ha evolucionado para suplir de nutrientes y oxígeno a las gruesas paredes miocárdicas que desde el lumen cardíaco y por difusión se produce en otras especies. Intervienen complejos y no del todo comprendidos mecanismos; una elaborada interacción de células epicárdicas con células miocárdicas llevan a la transformación de las primeras en células mesenquimales, precursoras de los bloques de construcción de los vasos coronarios8. Células progenitoras hematopoyéticas juegan un rol importante en la transformación de células epicárdicas en mesenquimales (Figura 2).
Desde el día 25 del desarrollo embrionario se identifican estructuras similvasculares, sin flujo, localizadas en el espacio entre epicardio y miocardio; en días posteriores estas estructuras se fusionan para formar plexos vasculares. Esta elaborada red finalmente se fusiona tomando contacto y uniéndose a la raíz aórtica, donde se expone a altos flujos y presiones sistémicas (Figura 3).
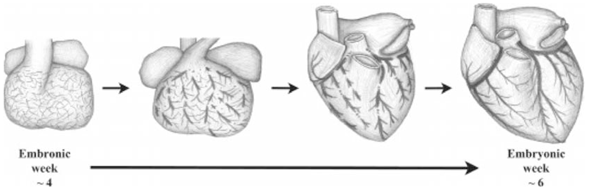
Estos cambios resultarán en la maduración de los vasos incluyendo migración de células musculares lisas, crecimiento de algunos vasos y regresión de otros mediante el mecanismo de apoptosis. Cualquier anomalía presente en este delicado y bien controlado proceso llevará al desarrollo de una anomalía congénita arterial coronaria8).
En la profundidad del epitelio dos procesos principales llevan a la formación de la vasculatura coronaria (vasculogénesis y angiogénesis) seguidos en el corazón adulto y en algunas instancias particulares de arteriogénesis (arterialización de colaterales preexistentes en áreas isquémicas)4.
Datos recientes sugieren que los vasos arteriales y venosos coronarios se originarían desde diferentes poblaciones de células embriogénicas endoteliales, desde diferentes sitios anatómicos y con diferencias temporales, y si bien el conocimiento actual que explica el desarrollo del sistema arterial coronario va en aumento hay poca información del desarrollo embriológico del sistema venoso cardíaco.
Históricamente se han definido y descrito las anomalías coronarias de acuerdo a elaborados sistemas de clasificación anatómica; básicamente las mismas varían entre:
Sin embargo, actualmente ha aumentado la necesidad de categorizarlas de acuerdo al significado pronóstico y más específicamente a la asociación con arritmias malignas o muerte súbita cardíaca. Las anomalías coronarias clínicamente significativas se clasifican en dos grupos con una base anatómica: aquellas con un origen en el seno aórtico opuesto y aquellas con un origen anómalo desde el tronco de la arteria pulmonar.
a) Arteria coronaria con origen en el seno opuesto; coronaria derecha naciendo en seno izquierdo o viceversa. De estas, la más frecuente es el nacimiento de la arteria circunfleja izquierda en el seno derecho. Las formas con curso interarterial (surco entre aorta y arteria pulmonar) aumentan el riesgo de compresión y consiguiente ocurrencia de arritmias malignas y muerte súbita que ocurren habitualmente en el ejercicio.
b) El origen coronario desde el tronco de arteria pulmonar es menos frecuente siendo el origen de la arteria coronaria izquierda la mayoría (ALCAPA). Se pueden presentar en etapas precoces de la vida con fallo cardíaco, miocardiopatía dilatada e insuficiencia mitral; el desarrollo de una red colateral e hipertensión pulmonar pueden dilatar el inicio de los síntomas.
Las anomalías en el curso mural de las arterias coronarias así como las fístulas coronarias o conexiones a cualquiera de las cámaras cardíacas o a grandes vasos escapan al objetivo del caso.
La presentación clínica no es uniforme. La fisiopatología de ARCAPA depende de la dirección del flujo sanguíneo en la arteria coronaria y su influencia en la entrega de oxígeno al miocardio, siendo la isquemia miocárdica determinada por el grado de shunt, el grado de circulación colateral y las demandas miocárdicas de oxígeno. Dado que las demandas a nivel ventricular derecho son menores que a izquierda la isquemia observada en ARCAPA es menos significativa. Esta situación no se cumple en aquellos pacientes con circulación coronaria derecha dominante1,6.
La cineangiocoronarioagrafía (CACG) representa el gold standard en el diagnóstico, aunque pueden existir complicaciones técnicas para la cateterización arterial. La angio-TAC con reconstrucción 3D y la resonancia nuclear magnética pueden ser consideradas al momento de estudiar el árbol coronario5,6,10. La ecocardiografía transtorácica, si bien con limitaciones, ha demostrado su rol importante en la evaluación de las anomalías coronarias principalmente en la evaluación del ostium y del primer segmento arterial dependiendo de la anatomía y ventana acústica. La técnica elegida debe ayudar a definir la presencia de otras anomalías estructurales cardíacas10.
Si bien no existen hallazgos electrocardiográficos característicos de ARCAPA, podemos encontrar hipertrofia ventricular izquierda con ondas Q profundas en cara inferior2,11,12.
Independiente de la caracterización anatómica, la terapia quirúrgica es la solución definitiva debido a la necesidad de establecer una circulación coronaria doble9. Dado el riesgo de isquemia o muerte súbita y de la mejora en la morbilidad incluso en pacientes asintomáticos luego de la corrección quirúrgica; el restablecer una circulación coronaria dual anterógrada mediante el reimplante de la arteria coronaria aberrante al anillo aórtico es recomendable como terapia definitiva2,6,5,10,11,13. La mortalidad operatoria se sitúa en 2%-3%1.
Conclusión
El desarrollo arterial coronario es un delicado y sincronizado proceso que incluye una compleja interacción entre pericardio, miocardio, múltiples vías celulares y diversos factores aún no completamente conocidos, siendo su estudio actual un proceso en crecimiento. Existen diferentes patrones de anomalías coronarias con variable riesgo de isquemia, arritmias o muerte súbita. El origen anómalo de la arteria coronaria derecha desde el tronco de la arteria pulmonar es una rara condición, con escasas manifestaciones clínicas y con evolución que puede llevar a un desenlace fatal. En nuestra paciente se realiza el diagnóstico ecocardiográfico en un estudio de rutina en el período neonatal, se confirma y determina su anatomía precisa mediante cateterismo cardíaco. Se realiza la corrección definitiva a los seis meses mediante reimplante coronario restableciendo la circulación coronaria dual.

