










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
Print version ISSN 0004-0584On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.87 no.2 Montevideo June 2016
Actualización de hepatitis autoinmune en pediatría. Reporte de un caso clínico y revisión de la literatura
Update on autoimmune hepatitis in pediatrics. Report of a clinical case and review of literature
Mariángel Ospitaleche1, Graciela Lagomarsino2, Catalina Pinchak3
1. Residente Pediatría. HCFFAA.
2. Pediatra. Gastroenteróloga. HCFFAA.
3. Prof. Agda. Clínica Pediátrica. Facultad de Medicina. UDELAR.
Hospital Central Fuerzas Armadas
Trabajo inédito.
Declaramos no tener conflictos de intereses.
Fecha recibido: 26 de febrero de 2014.
Fecha aprobado: 18 de marzo de 2016.
Resumen
La hepatitis autoinmune (HAI) es un proceso inflamatorio crónico y progresivo del hígado, de etiología desconocida. Se caracteriza por presentar niveles elevados de aminotransferasas e inmunoglobulina G (IgG), autoanticuerpos séricos y actividad necroinflamatoria en la histología; en ausencia de una patología conocida que pueda afectar al hígado. Predomina en el sexo femenino. Se describen dos tipos de acuerdo a los autoanticuerpos encontrados. El tratamiento se basa en la inmunosupresión, con el objetivo de evitar la progresión a cirrosis y falla hepática. Los pacientes no respondedores, o que debutan con falla hepática aguda pueden requerir de trasplante hepático. El objetivo es realizar una revisión del tema a partir de un caso clínico.
Se presenta a una adolescente de 14 años, derivada para estudio por probable patología autoinmune. El diagnóstico inicial fue de probable HAI, que se presentó como una falla hepática grave-fulminante. Posteriormente al descartar otras etiologías (infecciosas y metabólicas), presentar autoanticuerpos antinucleares (ANA) positivos y evidenciar cirrosis en la punción biópsica hepática, se confirmó el diagnóstico de cirrosis autoinmune. Se inició tratamiento con prednisona y azatioprina, con buena respuesta clínica y de laboratorio.
El diagnóstico oportuno de HAI y el inicio del tratamiento en forma temprana evitan en la mayoría de los casos la progresión de la enfermedad y el requerimiento de trasplante hepático.
Palabras clave:
HEPATITIS AUTOINMUNE
CIRROSIS HEPÁTICA
AZATIOPRINA
PREDNISONA
Summary
Autoimmune hepatitis is a chronic and progressive inflammatory process of the liver, of unknown etiology. It is characterized by presenting increased levels of aminotransferases and immunoglobulin G (IgG), serum antibodies and histologic necro-inflamatory activity, with unknown pathology that may affect the liver. It is more frequent in women, Two types of autoimmune hepatitis are described, according to the antibodies found. Treatment is based on immunosuppression, with the objective of avoiding progression to cirrhosis and liver failure.
Patients who do not respond or who present with severe liver failure may require liver transplant.
The objective of the study is to review the topic based on a clinical case.
The study presents the case of a 14 year old adolescent who is referred to be examined for possible autoimmune pathology. Initial diagnosis was probable autoimmune hepatitis, which presented with acute liver failure. Subsequently, when other etiologies were discarded (infectious and metabollic), positive antinuclear autoantibodies were present and liver hepatic biopsy evidenced cirrhosis, autoimmune cirrhosis was confirmed. Treatment was initiated with prednisone and azathioprine, being the clinical and lab response good.
In most cases, timely diagnosis of autoimmune hepatitis and early initiation of treatment avoid progression of the disease and the need for a liver transplant.
Key words:
AUTOIMMUNE HEPATITIS
LIVER CIRRHOSIS
AZATHIOPRINE
PREDNISONE
La HAI es un proceso inflamatorio crónico y progresivo del hígado de etiología desconocida, cuya patogenia se atribuye a una reacción inmune frente a autoantígenos hepatocelulares(1-3). Se caracteriza por elevación plasmática de las transaminasas, hipergammaglobulinemia, presencia de autoanticuerpos séricos hepáticos y actividad necroinflamatoria en la histología; en ausencia de una patología conocida que pueda afectar al hígado. Ocasionalmente puede manifestarse como una enfermedad multisistémica, asociándose a otros procesos autoinmunes(1-4).
Presenta una incidencia de 1-2/100.000 habitantes por año y una prevalencia de 11 a 17/100.000 habitantes. Predomina en el sexo femenino en la etapa prepuberal y puede presentarse en todas las razas, si bien es más prevalente en la raza blanca(1-4). La HAI es la tercera causa de trasplante hepático en Argentina luego de la atresia de vías biliares y de la falla hepática fulminante(5). No existen datos publicados a nivel nacional.
El diagnóstico se realiza en base a pilares clínicos, de laboratorio e histopatológicos. Se describen dos tipos de HAI de acuerdo a los autoanticuerpos plasmáticos encontrados.
La gravedad es variable. En los niños a menudo tienen un curso más agresivo que en los adultos. Pueden tener solo signos bioquímicos de disfunción hepática, lesiones propias de la hepatopatía crónica o presentar insuficiencia hepática aguda (10%)(5). El requisito antiguo de una duración de síntomas de 6 meses antes del diagnóstico ha sido abandonado(2).
El tratamiento se basa en la inmunosupresión, con el objetivo de evitar la progresión a cirrosis y falla hepática(1-9). Aquellos pacientes no respondedores o que debutan con falla hepática aguda pueden requerir de un trasplante hepático.
El objetivo de esta presentación fue realizar una revisión del tema a partir del reporte de un caso clínico.
Caso clínico
TM, 14 años. Sexo femenino. Antecedentes familiares: diabetes, patología tiroidea, vitíligo y abuela paterna fallecida por fallo hepático de etiología no filiada.
Buen crecimiento y desarrollo madurativo. Bien inmunizada, no recibió la vacuna contra hepatitis A. No antecedentes ambientales de hepatitis, no exposición a tóxicos ni ingesta de medicamentos.
Antecedentes patológicos: episodios de epistaxis leves en los últimos 4 meses.
Instala dolor abdominal 48 horas previas a la consulta, vómitos con sangre roja de escaso volumen y gingivorragias. Tránsito urinario: coluria. No fiebre. Se constató ictericia de piel y mucosas no evidenciada previamente. De los exámenes realizados se destaca: funcional hepático y crasis alterados, anemia normocítica y dosificación de T4 baja. Ecografía abdominal: esplenomegalia grado II. Valorado por cirujano interpreta: sangre deglutida por epistaxis. Se deriva a Montevideo para ampliar valoración diagnóstica y conducta terapéutica.
Al examen al ingreso: peso 61,9 kg; IMC: 23 (percentil 50-85). 36°C. Glasgow 15, sin cambios en el humor. Hemodinámicamente estable. Ictericia de piel y conjuntivas. No lesiones hemorragíparas. No circulación colateral. Examen abdominal: hígado firme y esplenomegalia grado II. Bucofaringe: múltiples focos sépticos dentarios.
Diagnóstico al ingreso: hepatitis de probable etiología autoinmune, que se presenta como una falla hepática grave, sin manifestación de encefalopatía portosistémica (EPS).
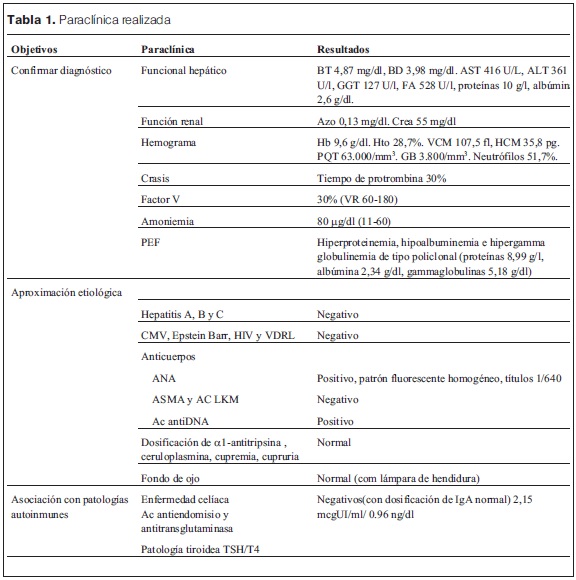
La paraclínica realizada se expone en la tabla 1.
La endoscopía digestiva descartó hipertensión portal (várices esofágicas, gástricas o gastropatía hipertensiva) como etiología del sangrado.
Se confirma el diagnóstico de hepatitis por signos clínicos y alteraciones en el funcional enzimograma hepático, presentando elementos de fallo hepático como son la alteración en el tiempo de protrombina e hipoalbuminemia. Por presentar ANA y anti DNA positivos, se planteó HAI tipo 1.
Se realizó tratamiento con vitamina K 10 mg i/v cada 12 h y transfusión de plasma.
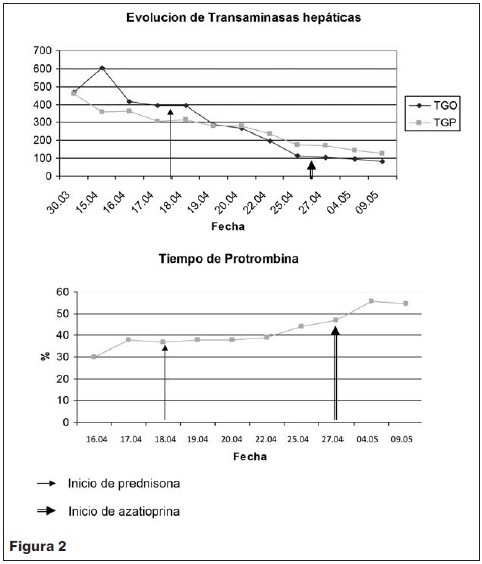
Al obtener los marcadores virales negativos se inició prednisona 40 mg por día v/o. Presentó buena respuesta clínica y de laboratorio (descenso progresivo de enzimas en el hepatograma) (anexo). Persiste con trastorno de la crasis por lo que al noveno día de tratamiento, con los anticuerpos positivos, se asoció azatioprina 50 mg por día v/o.
El puntaje de MELD al ingreso fue de 23, con lo cual se iniciaron los trámites correspondientes para su evaluación pretrasplante. Al momento del alta presentó un puntaje de 12.
La biopsia hepática se pospuso un mes por persistencia de trastornos en la crasis. Informe anatomopatológico: cirrosis en actividad compatible con etiología autoinmune. Por lo que se confirma el diagnóstico de cirrosis autoinmune y el de fallo hepático agudo sobre crónico en una paciente portadora de HAI.
Se disminuyó en forma gradual la dosis de prednisona v/o y se ajustó la azatioprina a 75 mg v/o por día. No tuvo efectos secundarios. En los controles posteriores se destaca la no adherencia al tratamiento y la presencia de conductas de riesgo.
En el caso presentado la impresión clínica inicial fue de una paciente portadora de hepatitis autoinmune, que se presentó con elementos de falla hepática grave. La presencia de esplenomegalia, con anemia, plaquetopenia y leucopenia en los exámenes sugirió una evolución crónica de la patología de base, que se confirmó con el hallazgo histopatológico de cirrosis autoinmune.
La presentación clínica de la HAI es proteiforme, desde pacientes asintomáticos a otros con un comienzo agudo, incluso fulminante (5%-10%). La forma de presentación más frecuente es como una hepatitis aguda viral(5).
En algunos casos el curso es insidioso y los pacientes presentan: fatiga, malestar general, cambios conductuales, anorexia, náuseas, dolor abdominal y amenorrea incluso varios meses antes de que se presente la ictericia o signos de hepatopatía crónica(1-5). El síntoma más frecuente es la astenia (86%) y el signo la hepatomegalia (78%)(9). En un número menor de pacientes, la clínica es sugestiva de cirrosis con edema, ascitis, várices hemorrágicas o EPS(4).
Manifestaciones extrahepáticas: artritis, vasculitis, nefritis, tiroiditis, anemia hemolítica y erupción cutánea(1).
Los mecanismos patogénicos de la HAI son desconocidos. La hipótesis más popular es que se trata de una etiología multifactorial. Existen evidencias que implican a los virus del sarampión, Epstein Barr, citomegalovirus y virus de la hepatitis A como agentes disparadores, probablemente por un mecanismo de mimetismo molecular(4,9). La predisposición genética (HLA-A1, B8, DR3, DR4, DR52a, DW3 y DRB1*1301) y varios determinantes de la presentación de autoantígenos, la activación de células inmunitarias y la expansión de células efectoras estarían involucrados(5,9-11).
La hipoalbuminemia es frecuente. La prolongación del tiempo de protrombina se debe la mayoría de las veces a déficit de vitamina K asociado al daño hepatocelular. En nuestra paciente se interpretó como disminución de la capacidad biosintética hepática por su cirrosis y en el caso que fuera menor que el basal (en este caso desconocido), como un elemento de descompensación de la hepatopatía.
Existe anemia normocítica, normocrómica, leucopenia y trombocitopenia, que se agrava cuando se desarrolla hipertensión portal e hiperesplenismo.
La detección por inmunofluorescencia indirecta (IFI) de ANA, autoanticuerpos antimúsculo liso (SMA) y anticuerpos antimicrosoma de hígado y riñón de rata tipo 1 (a-LKM1) constituye un criterio clave para el diagnóstico.
- Los ANA se determinan por IFI o ELISA. En niños se considera positivo ³1:20.
- Los SMA se dirigen frente a componentes de actina y no actina, son menos prevalentes que los ANA. En niños se considera positivo ³1:20. Su especificidad es mayor si son antiactina. Se asocian al haplotipo HLA DR3 y tienen carácter pronóstico (peor respuesta al tratamiento).
- Los a-LKM van dirigidos contra el citocromo P4502D6. Se considera positivo valores ³1:10.
- Los anticuerpos anti-DNA suelen asociarse con el haplotipo HLA DR4 e identifica pacientes con peor respuesta al tratamiento corticoideo.
Hasta un 20% de los pacientes con probable HAI pueden no tener autoanticuerpos al momento de la presentación.
Otros anticuerpos que actualmente no se encuentran disponibles en nuestro país son:
- Anti-ASGRP: su persistencia anuncia recidivas tras la retirada de corticoides.
- Anticuerpos frente a antígeno soluble hepático y antihígado páncreas (anti-SLA/LP): están presentes en el 50% de los tipos 1 y 2 y definen un curso más severo. Se asocia al HLA DR3 y predisposición a recaer(11).
- Anti-LC1: es específico de HAI y su diana antigénica es la formiminotransferasa y la argininosuccinatoliasa. Están presentes también en pacientes con colangitis esclerosante primaria. Sus niveles fluctúan con la actividad inflamatoria y pueden ser útiles como marcadores de inflamación hepatocelular.
Se describen dos tipos de HAI de acuerdo a los autoanticuerpos circulantes.
- HAI tipo 1: 80% de los casos(2,11). Es más prevalente en mujeres (2:1), 40% en la edad pediátrica. La media de edad de presentación: 10 años. Se caracteriza por la presencia de ANA y/o SMA. Hasta en el 60% pueden detectarse autoanticuerpos frente a antígeno SLA/LP y en menor frecuencia anticuerpos antimitocondriales (AMA, típicos de la cirrosis biliar primaria) y pANCA atípicos(1). Se asocia a un inicio brusco de los síntomas en el 40% de los casos y puede tener presentación fulminante. Los pacientes con una presentación aguda muestran cambios clínicos, paraclínicos e histológicos que hacen sospechar de una hepatopatía crónica subyacente, como sucedió en nuestra paciente. Patologías autoinmunes asociadas (41%): hipertiroidismo (12% tiroiditis autoinmunitaria y 6% enfermedad de Graves), colitis ulcerosa crónica (6%) y anemia hemolítica(9).
- HAI tipo 2: 20% de los casos. Predomina en preadolescentes y mujeres jóvenes (6:1), 80% en la edad pediátrica. La media de edad de presentación: 6,5 años. Presenta autoanticuerpos LKM. Con frecuencia la presentación es aguda, a veces fulminante, con tendencia a progresar rápidamente a cirrosis y es la de peor pronóstico. Patologías autoinmunes asociadas: diabetes mellitus tipo 1, enfermedad tiroidea y vitíligo. Puede ser parte del “autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED)”, un desorden autosómico recesivo en donde la lesión hepática se encuentra en un 20% de los casos(11).
El diagnóstico de HAI tipo 3 ha sido abandonado, dado que su marcador, el antígeno SLA/LP, también se ha encontrado en la HAI tipo 1 y 2(2).
Los pacientes con HAI de características atípicas no cuentan con una designación oficial. Pueden tener manifestaciones de HAI y de otros tipos de hepatopatía crónica (síndrome de solapamiento) o signos que son incompatibles con la HAI según los criterios diagnósticos actuales (síndrome fuera de rango)(1,4,6,9). La HAI que también tiene anticuerpos AMA o falta de respuesta al tratamiento corticoideo y características histológicas de colangitis constituyen un síndrome de solapamiento con la colangitis esclerosante. En estos casos se justifica la realización de una colangiografía.
Diagnóstico de HAI
Es de exclusión, luego de evaluar otras causas de lesión hepatocelular crónica: infecciones por virus hepatotropos como el virus de hepatitis B, C y D (este último como coadyuvante del VHB en la injuria hepática), hepatopatías por fármacos y tóxicos o por depósito (hemocromatosis, enfermedad de Wilson), o la deficiencia de a1-antitripsina.
Se debe realizar consulta con especialista en Gastroenterología quien ampliara valoración diagnóstica, realizará seguimiento y tratamiento del paciente
Los criterios clínicos son suficientes para diagnosticar una HAI definitiva o probable en la mayoría de pacientes, como en la que presentamos (tabla 2).

En los casos difíciles se puede aplicar el sistema de puntuación diagnóstico. El primero utilizado fue definido por el Grupo Internacional de HAI (IAIHG) en 1992 modificado por Brighton en 1999 (tabla 3).
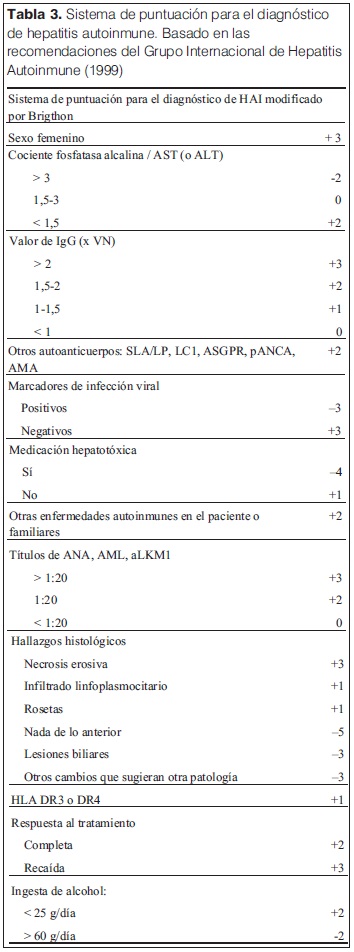
En los pacientes no tratados se considera que el diagnóstico es definitivo con puntuación >15, probable 10-15 y excluyente < a 10. Nuestra paciente presentó 19 puntos. En los pacientes que reciben tratamiento inmunosupresor, hay certeza diagnóstica si es >17 y probable de 12-17puntos.
Una puntuación de 10 puntos antes del tratamiento tiene una sensibilidad de 100%, especificidad 73% y precisión diagnóstica de 67%. Un puntaje pretratamiento de 15 puntos tiene una sensibilidad de 95%, especificidad 97% y una precisión diagnóstica de 94%.
En 2008 la Internacional Autoinmune Hepatitis Group (IAIHG) publicó un score más simple basado en anticuerpos, IgG, histología y exclusión de hepatitis virales que permiten una mejor aplicación clínica (tabla 4)(13,14).
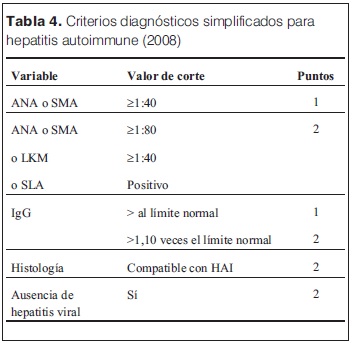
Con score ³6 se identifica probable HAI, y ³7 es definitivo. La sensibilidad es de 88% y la especificidad del 97% para casos probables, de 81% y 99% para casos definitivos respectivamente.
Ninguno de los scores es perfecto en particular en pediatría, en donde los puntos de corte para los anticuerpos son mas bajos. Ninguno de los dos puede distinguir la HAI de la colangitis esclerosante autoinmune(14,15). La positividad para anticuerpos no es suficiente para el diagnóstico, dado que pueden encontrarse títulos bajos en pacientes con hepatitis virales, enfermedad de Wilson y esteatosis hepática de causa no alcohólica(11).
Las guías de la Asociación Americana para el Estudio de Enfermedades Hepáticas (AASLD, por su sigla en inglés) en 2010 sugieren las consideraciones siguientes:
1. El diagnóstico debe ser realizado en pacientes con clínica, alteraciones de laboratorio e histológicas compatibles e incluyen pruebas bioquímicas hepáticas anormales, un aumento de IgG total o niveles de gamma-globulinas, marcadores serológicos (ANA, SMA, anti-LKM-1, o anti-LC1) y hallazgos histopatológicos de hepatitis de interfase.
3. En casos dudosos, se debe usar el sistema de puntuación estandarizado.
4. En pacientes con autoanticuerpos convencionales negativos, se debe buscar anticuerpos adicionales (mínimo incluir anti-SLA y pANCA)(2,8).
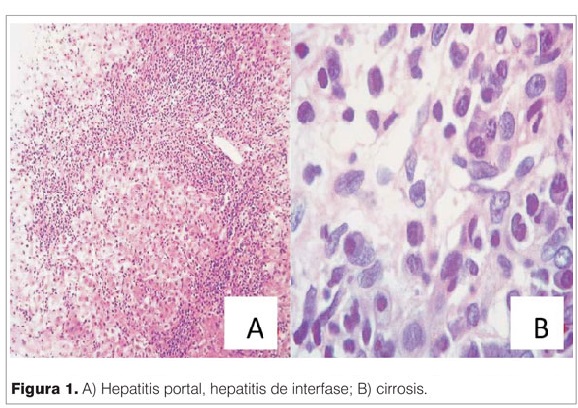
La biopsia hepática es un pilar diagnóstico, permite definir estadio y actividad. La HAI suele ser un trastorno primario y específico del hepatocito. En los niños puede ser común la superposición de hallazgos que afectan tanto al hepatocito como el conducto biliar(4). Los hallazgos no son específicos. Las características histológicas típicas consisten en un infiltrado denso de células mononucleares (linfocitos T, B, macrófagos y células plasmáticas) portales, que invade el parénquima circundante y a menudo penetra en el lobulillo (hepatitis de interfase). Puede observarse necrosis parcheada de los hepatocitos de moderada a grave, varios grados de necrosis, fibrosis y zonas de colapso del parénquima que engloban tríadas portales adyacentes o situadas entre una tríada portal y una vena central (necrosis en puente) y grados variables de lesión del epitelio del conducto biliar(16). En 50% de los casos se constata cirrosis al momento del diagnóstico, como en nuestro caso (figura 1).
La biopsia hepática forma parte del algoritmo de estudio de las enfermedades hepáticas pediátricas. La secuenciación del ADN, nuevas modalidades de imagen, la elastografía de transición hepática (método biofísico no invasivo de detección de fibrosis), la proteómica, y estudios de asociación del genoma ofrecen posibles métodos alternativos en el futuro para la evaluación de la enfermedad hepática.
Debe iniciarse tempranamente en el paciente con evidencia de hepatopatía autoinmune, una vez descartada la etiología infecciosa y metabólica.
Los objetivos son: aliviar la sintomatología, mejorar los indicadores bioquímicos, suprimir o eliminar la inflamación hepática y disminuir la mortalidad con los mínimos efectos secundarios(2).
Las guías de la AASLD establecen las indicaciones absolutas de tratamiento de acuerdo a los niveles de aminotransferasas, IgG y los hallazgos histopatológicos. Sin embargo considera que todos los niños deben recibir tratamiento al momento del diagnóstico(2).
Existe consenso respecto a que el tratamiento de elección al inicio es con prednisona 1-2 mg/kg/día (máximo 60 mg/día), hasta que los valores de aminotransferasas descienden a menos del doble de lo normal, tras lo cual se reduce la dosis de 5 en 5 mg hasta una dosis de mantenimiento de 0,1-0,2 mg/kg/día. En la mayoría de los pacientes se alcanza una reducción de 80% de los valores de aminotransferasas en los primeros 2 meses, el valor normal puede tardar varios meses en alcanzarse(4,11). Se debe asociar vitamina D y calcio como medidas profilácticas de enfermedad ósea.
La asociación de prednisona con azatioprina mejora las características clínicas, bioquímicas e histológicas en la mayoría de los pacientes con HAI y prolonga su supervivencia(2,17,18).
La azatioprina es un antagonista de las purinas que bloquea la proliferación de linfocitos. Dosis inicial: 1,5-2 mg/kg/día, aumentando en forma progresiva. No es eficaz como monoterapia para inducir la remisión, pero sí ha demostrado beneficios en mantener la remisión, minimizando los efectos adversos de los corticoides(17). El tiempo de inicio de la asociación varía de acuerdo a protocolos realizados en cada centro, algunos después de unas pocas semanas con corticoides y otros desde el comienzo(2,18). Los efectos secundarios son: depresión de la médula ósea, pancreatitis aguda, hepatotoxicidad colestásica con fibrosis y mayor predisposición a infecciones(2,18).
En nuestra paciente se inició tratamiento con prednisona y a los nueve días se asoció azatioprina con mejoría clínica progresiva, buena tolerancia y tendencia a la normalización del laboratorio.
Se ha asociado ácido ursodesoxicólico al tratamiento inmunosupresor cuando existen evidencias de colestasis. Dosis: 13-15 mg/kg/día(19).
El 70%-80% de los pacientes responden adecuadamente al tratamiento inmunosupresor. Éste mejora la supervivencia, con una esperanza de vida a los 10 años después del inicio del mismo en los pacientes con y sin cirrosis en el momento del diagnóstico del 89% y 90% respectivamente.
Remisión, recaída y fracaso al tratamiento
Remisión: recuperación clínica completa, con valores normales de transaminasas e IgG, títulos bajos o negativos de anticuerpos y resolución histológica de la inflamación(19,20).
Fracaso al tratamiento: aumento de los niveles de aminotransferasas o bilirrubina al menos en un 67% de los valores previos, actividad histológica progresiva o inicio de ascitis o signos de encefalopatía portosistémica.
Otras alternativas terapéuticas existentes serán valoradas por el gastroenterólogo tratante en el caso de presentaciones atípicas o de que la evolución no sea la esperada: budesonida, ciclosporina, tacrolimus (FK-506), micofenolato de mofetilo y sirolimus(1-3).
La duración óptima del tratamiento inmunosupresor es desconocida. Es aconsejable no retirar el tratamiento antes de los 2 años de realizado el diagnóstico o durante la etapa prepuberal inmediata, dado que las recaídas son más frecuentes(23). Solo en 20% de los casos de HAI tipo 1 se logra la suspensión del tratamiento y raramente en los de tipo 2.
En un paciente con fallo hepático grave o que no responde al tratamiento inmunosupresor se exige la valoración temprana de indicación de trasplante hepático, como se realizó en nuestra paciente.
El trasplante hepático ortotópico ha tenido éxito en pacientes con hepatopatía terminal asociada a HAI. La supervivencia de los pacientes y de los injertos varía entre 83% y 92% respectivamente y la tasa de supervivencia a los 10 años del trasplante es del 75%. La indicación de trasplante se debe basar en la existencia de descompensación hepática(24). Los pacientes con fallo hepático al debut presentan refractariedad al tratamiento en el 50%-75% de los casos, necesitando del trasplante para su sobrevida.
El Model for End-Stage Liver Diseas (MELD) es un índice pronóstico de mortalidad utilizado para valorar la gravedad de la hepatopatía, objetivo y fácilmente reproducible, basado en tres variables analíticas simples: la bilirrubina, la creatinina sérica y el INR. Se correlaciona muy bien con la mortalidad a tres meses y desde el año 2002 se utiliza para priorizar a los pacientes mayores de 12 años en lista de espera de trasplante hepático con un puntaje mayor a 14.
El fracaso del tratamiento se predice mejor por el cambio en el MELD en el día 7. La identificación temprana de los pacientes que no responden a tiempo puede permitir la escalada de la inmunosupresión para evitar el deterioro clínico.
El Pediatric Liver Disease Severity Score (PELD) es utilizado en menores de 12 años con el mismo objetivo que el MELD, pero a diferencia de este último no usa el valor de la creatinina y si incluye albúmina, existencia o no de déficits del crecimiento y la edad(25).
En el caso de un paciente con indicación de trasplante hepático, se debe descargar de la página web http://www.fnr.gub.uy/ el formulario de evaluación pretrasplante del Fondo Nacional de Recursos (FNR). El pediatra o gastroenterólogo tratante deben completarlo y presentarlo ante el FNR en la dirección Avda. 18 de Julio 985, Galería Cristal 3er. piso. Tel: 2901 4091. Luego de la evaluación por la comisión técnico administrativa se notifican a los padres y se genera la primera evaluación por hepatólogos.
La unidad integrada por el Hospital Central de las Fuerzas Armadas se encarga del seguimiento y control de los pacientes pediátricos en lista de espera y de los ya trasplantados que pertenecen a todo el sistema integrado de salud. Desde julio de 2009, el trasplante hepático en adultos se realiza en Uruguay.
La recaída de la enfermedad postrasplante (25%-50% de niños) es más frecuentes que en adultos y más grave, condicionando la pérdida de injertos frecuentemente(26).
Factores de mal pronóstico
Menor de 2 años de edad, tiempo de protrombina alargado (>62 seg, INR >4), cifras altas de bilirrubina (>13,7 mg/dl), anticuerpos LKM positivos, índice alto de actividad histológica o cirrosis y los fenotipos HLA-B8 y HLA-DR3(1-6).
El seguimiento debe realizarse por pediatra y gastroenterólogo. Se basa en controles clínicos y paraclínicos (hemograma, funcional-enzimograma hepático y amilasa) cada 4 semanas hasta la remisión, realizándose luego de manera trimestral (depende de los empujes y remisiones de la enfermedad)(1). Deben realizarse controles oftalmológicos para descartar la aparición de cataratas.
Dado que la hepatitis por virus A puede potencialmente agravar la patología preexistente se debería inmunizar a la paciente en cuanto sea posible.
Referencias bibliográfícas
1. Galicia G, Manzanarez J. Hepatitis autoinmune. En: Asociación Española de Pediatría. Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica. Protocolos diagnósticos terapéuticos de gastroenterología, hepatología y nutrición Pediátrica. Madrid: Ergón, 2010:211-20.
2. Manns MP, Czaja A, Gorham J, Krawitt E, Mieli-Vergani G, Vergani D, et al; American Association for the Study of Liver Diseases. Diagnosis and management of autoimmune hepatitis. Hepatology 2010; 51(6):2193-213.
3. Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica. Tratamiento en gastroenterología, hepatología y nutrición pediátrica. Madrid: Ergón, 2008.
4. Shneider B, Suchy F. Hepatitis autoinmunitaria. En: Kliegman M, Stanton F, Gemell W, Schor F, Behrman E. Nelson tratado de pediatría. 19 ed. Barcelona: Elsevier, 2013:1464-7.
5. Cuarterolo M, Ciocca M, Alvarez F. Hepatitis autoinmune en niños: perspectivas actuales. Arch Argent Pediatr 2014; 112(2):169-75.
6. Moreno R, García-Monzon C, García-Buey L. Hepatitis autoinmune. En: Asociación Española para el Estudio del Hígado. Tratamiento de las enfermedades hepáticas y biliares. 2 ed. Madrid: AEEH, 2001:63-73.
7. Duarte-Rey C, Pardo AL, Rodríguez-Velosa Y, Mantilla R, Anaya JM, Rojas-Villarraga A. HLA class II association with autoimmune hepatitis in Latin America: a meta-analysis. Autoimmun Rev 2009; 8(4):325-31.
8. Prieto J, Preciado J, Huertas S. Hepatitis autoinmune. Rev Col Gastroenterol 2012; 27(4):303- 15. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0120- 99572012000400007. [Consulta: 6 enero 2013].
9. Feldman M, Friedman L, Sleisenger MH. Eds. Sleisenger & Fordtran enfermedades gastrointestinales y hepáticas: fisiopatología, diagnóstico y tratamiento. Tomo 1. 7 ed. Buenos Aires: Médica Panamericana, 2004:1869-83.
10. Sotelo N. Diagnóstico y tratamiento de niños con falla hepática aguda. Rev Mex Pediatr 2010; 77(2):72-7.
11. Mieli-Vergani G, Vergani D. Is there a “Standard of care” in the diagnosis and treatment of pediatric autoinmune hepatitis? En: The 62nd Annual Meeting of the American Association for the Study of Liver Diseases. San Francisco, CA, November 5-8, 2011.
12. França R, Bittencourt P, Freitas L, May D, Andrade C, Cunha L, et al. Frequency of celiac disease and its serological markers in patients with autoimmune hepatitis. Rev Ciênc Méd Biol 2007;6(2):175-82.
13. Ferri P, Ferreira A, Miranda D, Simões A. Diagnostic criteria for autoimmune hepatitis in children: a challenge for pediatric hepatologists. World J Gastroenterol 2012; 18(33):4470-3.
14. Mileti E, Rosenthal P, Peters MG. Validation and modification of simplified diagnostic criteria for autoimmune hepatitis in children. Clin Gastroenterol Hepatol 2012; 10(4):e1-2. 417-21.
16. Ovchinsky N, Moreira R, Lefkowitch J, Lavine J. Liver biopsy in modern clinical practice: a pediatric point-of-view. Adv Anat Pathol 2012; 19(4):250-62.
17. Della C, Sartorelli MR, Sindoni C, Girolami E, Giovannelli L, Comparcola D, et al. Autoimmune hepatitis in children: an overview of the disease focusing on current therapies. Eur J Gastroenterol Hepatol 2012; 24(7):739-46.
18. Rumbo C, Emerick K, Emre S, Shneider B. Azathioprine metabolite measurements in the treatment of autoimmune hepatitis in pediatric patients: a preliminary report. J Pediatr Gastroenterol Nutr 2002; 35(3):391-8.
19. Vergani D, Mieli-Vergani G. Pharmacological management of autoimmune hepatitis. Expert Opin Pharmacother 2011; 12(4):607-13.
20. Trivedi P, Hirschfield G. Treatment of autoimmune liver disease: current and future therapeutic options. Ther Adv Chronic Dis 2013; 4(3):119-41.
21. Kerkar N, Annunziato R, Foley L, Schmeidler J, Rumbo C, Emre S, et al. Prospective analysis of nonadherence in autoimmune hepatitis: a common problem. J Pediatr Gastroenterol Nutr 2006;43(5):629-34.
22. Samaroo B, Samyn M, Buchanan C, Mieli-Vergani G. Long-term daily oral treatment with prednisolone in children with autoimmune liver disease does not affect final adult height. Hepatology 2006; 44: A438.
23. Cuarterolo ML, Ciocca ME, López S, de Dávila MT, Alvarez F. Immunosuppressive therapy allows recovery from liver failure in children with autoimmune hepatitis. Clin Gastroenterol Hepatol 2011; 9(2):145-9.
24. Martín S, Alvarez F, Anand R, Song C, Yin W; SPLIT Research Group. Outcomes in children who underwent transplantation for autoimmune hepatitis. Liver Transpl 2011; 17(4):393-401.
25. Wiesner R, McDiarmid S, Kamath P, Edwards E, Malinchoc M, Kremers W, et al. MELD and PELD: application of survival models to liver allocation. Liver Transpl 2001; 7(7):567-80.
26. Hadžiæ N, Quaglia A, Cotoi C, Hussain MJ, Brown N, Vergani D, et al. Immunohistochemical phenotyping of the inflammatory infiltrate in de novo autoimmune hepatitis after liver transplantation in children. Pediatr Transplant 2012; 16(5):501-10.
Correspondencia: Dra. Mariángel Ospitaleche.
Correo electrónico: mor2747@hotmail.com

