










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versão On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.86 no.3 Montevideo set. 2015
Síndrome de Guillain-Barré
Experiencia de doce años
Guillain-Barré syndrome. Twelve years of experience
Lucía Cibils1, Alfredo Cerisola2, Gabriela Capote3, Carina Ferreira4, Natalia Rodríguez4,
Conrado Medici5, Gabriel González6, Cristina Scavone7
1. Médico Residente Neuropediatría.
2. Prof. Agdo. Neuropediatría.
3. Médico Pediatra, Neuropediatra.
4. Médico Pediatra.
5. Asistente Cátedra Neuropediatría.
6. Prof. Director Cátedra Neuropediatría.
7. Prof. Neuropediatría. Ex–Directora Cátedra Neuropediatría.
Cátedra Neuropediatría, Facultad de Medicina, UDELAR. CHPR. ASSE.
Trabajo inédito.
Declaramos no tener conflictos de intereses.
Fecha recibido: 24 de noviembre de 2014.
Fecha aprobado: 26 de junio de 2015.
Resumen
Introducción: el diagnóstico y el tratamiento temprano del síndrome de Guillain-Barré (SGB) es importante para reducir la morbimortalidad de la enfermedad, por lo cual es capital conocer las manifestaciones clínicas iniciales y el rendimiento de las pruebas diagnósticas.
Objetivos: describir las características de los pacientes menores de 15 años hospitalizados por SGB en el Centro Hospitalario Pereira Rossell entre el 1 de enero de 2000 al 31 de diciembre de 2011.
Método: estudio descriptivo, retrospectivo, en base a revisión de historias clínicas.
Resultados: se incluyeron 82 pacientes, 52 eran de sexo masculino. La mediana de edad fue de 6 años. 59 pacientes requirieron internación en unidad de cuidados intensivos. El síntoma más frecuente fue la paresia de miembros inferiores (80 pacientes), seguido del dolor en miembros inferiores (64 pacientes). El 29% de nuestra serie se presentó clínicamente como variantes atípicas de SGB. La mitad de los pacientes presentó un Hughes 4 en el nadir de la enfermedad. A 77 pacientes se les realizó estudio del líquido cefalorraquídeo, reiterándose en 17 pacientes, mostrando disociación albúmino citológica en 19/53 (36%) de los estudios realizados en los primeros 7 días de evolución, y en 39/41 (95%) de los estudios realizados luego de los 7 días.
Conclusiones: se destacan las diversas manifestaciones clínicas del SGB y la baja sensibilidad del estudio del líquido cefalorraquídeo en la primera semana de evolución.
Palabras clave:
SÍNDROME DE GUILLAIN-BARRÉ
POLIRRADICULONEUROPATÍA
NEUROPATÍA MOTORA AGUDA
Introduction: early diagnosis and treatment of Guillain-Barré is important to reduce morbimortality of the condition and thus, it is essential to learn about initial clinical presentations and the yield of diagnostic tests.
Objectives: to describe the characteristics of patients under 15 years of age who were hospitalized due to Guillain-Barré syndrome at the Pereira Rossell Hospital Center from January 1, 2000 through December 31, 2011.
Method: descriptive, retrospective study based on a review of the clinical records.
Results: eighty two patients were included, 52 of them were male. Fifty nine patients required to be admitted to the intensive care unit. The most frequent symptom was paresia of the lower limbs (80 patients), followed by pain in the lower limbs (64 patients). Twenty nine percent of our series evidenced clinical presentations that were not typical of Guillain-Barré syndrome. Half of the patients presented disease severity at nadir grade 4 by Hughes. Seventy seven patients underwent a study of the spinal fluid, and this was repeated in 17 patients, what showed albuminocytologic dissociation in 19 out of 53 (36%) of the tests performed in the first seven days of evolution, and in 39 out of 41 (95%) of the tests performed after seven days.
Conclusions: we point out the diverse clinical presentations of the Guillain-Barré syndrome and the low sensitivity of the spinal fluid in the first week of evolution.
Key-words:
GUILLAIN-BARRE SYNDROME
POLYRADICULONEUROPATHY
ACUTE MOTOR NEUROPATHY
Introducción
Clásicamente, el síndrome de Guillain-Barré (SGB) se define como una polirradiculoneuropatía aguda, caracterizada por una parálisis flácida ascendente, simétrica y arrefléctica, que puede asociarse a síntomas sensitivos y alteraciones autonómicas. Con el avance de la investigación médica, el término de SGB comenzó a aplicarse a un amplio espectro de polirradiculoneuropatías inflamatorias agudas adquiridas, mediadas inmunológicamente, presumiblemente desencadenadas por una infección previa (postinfecciosas), con variados mecanismos fisiopatológicos (desmielinización, lesión axonal motora o sensitivo-motora) con manifestaciones clínicas diversas (paresia, arreflexia, ataxia, síntomas sensitivos, alteraciones autonómicas), con formas de presentación y grados de gravedad variables, y con una evolución generalmente monofásica y autolimitada(1,2). Existen formas graves con riesgo vital que requieren tratamientos en las unidades de cuidados intensivos y algunos pacientes quedan con secuelas motoras con diferentes grados de discapacidad(3). El diagnóstico se basa en los criterios de Asbury y Cornblath modificados que incluyen criterios clínicos, la presencia de disociación albúmino-citológica en el líquido cefalorraquídeo y hallazgos electrofisiológicos típicos(4-6). Los criterios electrofisiológicos de SGB y de sus variantes han sido bien definidos(7).
Es la causa más frecuente de parálisis flácida aguda en los países con programas de inmunización establecidos, donde la poliomielitis ha sido virtualmente erradicada(8,9). En los últimos años se ha reconocido que el SGB no puede considerarse una enfermedad única, sino que puede subdividirse en como mínimo cuatro subtipos diferentes con características electrofisiológicas, inmunopatogénicas y anatomopatológicas específicas: la polineuropatía inflamatoria desmielinizante aguda (PDIA), la polineuropatía axonal motora aguda (PAMA), la polineuropatía axonal sensitivo-motora aguda (PASMA) y el síndrome de Miller-Fisher (SMF). Asimismo, se han descrito múltiples variantes y formas intermedias(7,10). El problema no es el diagnóstico de los casos típicos de SGB, sino reconocer las fronteras que delimitan este trastorno y las formas de presentación atípica.
El diagnóstico se basa en los criterios de Asbury y Cornblath modificados establecidos en 1978 y modificados en 1990(11). Incluyen criterios clínicos, la presencia de disociación albúmino-citológica en el líquido cefalorraquídeo y hallazgos electrofisiológicos típicos(5,6). Dentro de los criterios clínicos se definen síntomas y signos requeridos para el diagnóstico (debilidad muscular progresiva de más de una extremidad e hipo o arreflexia). Apoyan el diagnóstico: una progresión de los síntomas de hasta cuatro semanas, simetría relativa, signos o síntomas sensitivos leves, compromiso de pares craneales, presencia de disfunción autonómica, dolor, ausencia de fiebre al comienzo de los síntomas y recuperación que generalmente comienza de dos a cuatro semanas luego de haber alcanzado la máxima gravedad.
El hallazgo característico en el LCR es la disociación albúmino-citológica. Este hallazgo aparece después de la primera semana de inicio del cuadro. Se admiten como variantes la falta de aumento de proteínas en el período de 1 a 10 semanas después del inicio de los síntomas y la presencia de 11 a 50 mononucleares/mm3 en el LCR.
La importancia del diagnóstico del SGB en un paciente con una paresia rápidamente progresiva radica en que el tratamiento con inmunoglobulinas intravenosas o plasmaféresis puede modificar la evolución de la enfermedad y disminuir la morbilidad y la mortalidad(2). Sin embargo, con frecuencia se plantean dificultades para formular el diagnóstico temprano del SGB. En primer lugar, los criterios diagnósticos del SGB no abarcan el espectro completo de este trastorno(14). En segundo lugar, los síntomas iniciales pueden ser difíciles de interpretar. Entre el 50%-80% de los niños tendrán dolor significativo al inicio del cuadro. En los niños pequeños, distinguir entre paresia muscular y la inmovilidad por dolor puede ser casi imposible. Por otro lado, casi la mitad de los pacientes se presentan con inestabilidad en la marcha, pero ésta puede deberse a debilidad muscular proximal o, en otros casos, a la disfunción de las vías sensitivas (ataxia sensitiva)(5). El diagnóstico también puede ser difícil en algunos niños que se presentan con cefaleas intensas, irritabilidad o somnolencia, lo que constituye una forma “pseudoencefalopática”, asociada o no a un dolor paraespinal con meningismo, que puede simular una meningitis aguda, una encefalitis o un tumor cerebral(5,11). Existen muchas otras formas de presentación atípicas que también responderán al tratamiento, pero si se amplía excesivamente la definición del SGB, pueden no considerarse otros diagnósticos diferenciales de enfermedades que, si no se reconocen oportunamente, pueden resultar en un diagnóstico tardío(5).
Las variantes atípicas incluyen formas de presentación clínica asimétricas, motoras puras, con déficit sensitivos predominantes, con preservación de los reflejos de estiramiento muscular o, incluso, hiperreflexia y con paresia bilateral inicial de los miembros superiores o comienzo en los nervios craneales y parálisis descendente(2,6,15,16). Otras formas atípicas son las variantes regionales de la PDIA, cuyas descripciones incluyen la parálisis faringocervicobraquial (2,6,14), la polineuropatía craneal múltiple(17), la diplejía facial con parestesias, la paresia del sexto par con parestesias, la ptosis palpebral grave sin oftalmoplejía, la paraparesia arrefléxica y la variante “saltatoria”, con afectación de los miembros inferiores y los pares craneales sin afectación de los miembros superiores(2,6). El SMF puede presentarse en su forma típica, con la tríada clásica de oftalmoparesia, ataxia y arreflexia, o como un síndrome superpuesto (compartiendo características de otras variantes de SGB) o como variantes más limitadas(1,6,18). Además se describen otros tipos de variantes, entre las que se incluyen las que presentan alteraciones de la oculomotricidad intrínseca (midriasis, enlentecimiento del reflejo fotomotor) o las que asocian una afectación del tronco cerebral (encefalitis benigna de tronco cerebral de Bickerstaff) (19). Otras variantes del SGB incluyen la neuropatía sensitiva pura, la disautonomía autonómica aguda pura y el SGB asociado con manifestaciones del sistema nervioso central (SNC)(20).
Con relación a los pilares paraclínicos que apoyan el diagnóstico del SGB, las dificultades en el diagnóstico se plantean, en primer lugar, por la falta de marcadores serológicos confiables del SGB. En segundo lugar, el estudio del LCR puede no ser diagnóstico hasta una o dos semanas después del inicio de la enfermedad, cuando hasta 80%-90% de los casos presentan una disociación albúmina-citológica(6). En tercer lugar, los estudios electrofisiológicos pueden ser difíciles de realizar en los niños pequeños y los hallazgos clásicos de desmielinización en los estudios de conducción nerviosa pueden no aparecer hasta dos o cuatro semanas después del inicio de la enfermedad(2,11), sumada a la dificultad en la clasificación de tipo de variante especialmente al inicio de la enfermedad, mencionada anteriormente.
Debido a todos estos factores, es frecuente que en los pacientes con SGB se formulen múltiples diagnósticos diferenciales en función de la forma de presentación inicial de cada caso. Entre éstos se incluyen las encefalopatías agudas en las formas pseudoencefalopáticas, las otras causas de ataxias(5), las mielopatías agudas (compresión de la médula espinal, mielitis transversa, encefalomielitis difusa aguda o infarto del territorio de la arteria espinal anterior)(5), las afecciones del asta anterior de la médula espinal (poliomielitis por virus nativo en áreas endémicas, por virus vacunal atenuado, por otros enterovirus o por virus del Nilo del Oeste), los otros tipos de neuropatías periféricas(2,5,6,10,21,22), las alteraciones de la unión neuromuscular (botulismo y miastenia gravis)(2,5,6) y las enfermedades musculares (miositis agudas inflamatorias, miopatías metabólicas, parálisis periódicas, con hiper o hipokaliemia o miopatías del paciente crítico(1,6)).
Objetivos
Objetivo general
- Describir las formas de presentación de los pacientes menores de 15 años con SGB y analizar las manifestaciones clínicas iniciales, su progresión y los estudios complementarios realizados.
Objetivos específicos
- Describir las características demográficas y epidemiológicas de los pacientes con SGB hospitalizados en el Centro Hospitalario Pereira Rossell (CHPR) durante un período de doce años.
- Describir el tiempo de evolución desde el ingreso hospitalario, los síntomas iniciales, las manifestaciones clínicas en toda la evolución y su forma de progresión e identificar las diferentes formas de presentación, en especial las variantes atípicas e inusuales, para valorar la heterogeneidad clínica y las dificultades que pueden plantearse en el diagnóstico del SGB.
- Describir los resultados de los estudios del LCR para apoyar el diagnóstico del SGB. Calcular y comparar la sensibilidad de esta prueba diagnóstica cuando se realiza en la primera semana de evolución y cuando se realiza después de la primera semana.
- Identificar los diagnósticos diferenciales registrados en las historias clínicas y los estudios complementarios realizados para valorar o descartar dichas hipótesis diagnósticas.
Pacientes y métodos
Estudio descriptivo y retrospectivo sobre la base de la revisión de las historias clínicas de una serie consecutiva de casos de niños y adolescentes menores de 15 años que ingresaron en el Hospital Pediátrico del CHPR por un SGB entre el 1 de enero de 2000 y el 31 de diciembre de 2011.
Se incluyeron aquellos pacientes con diagnóstico de SGB identificados en el análisis de las bases de datos existentes en el CHPR y cuyas características clínicas y paraclínicas coincidían con los criterios de Asbury y colaboradores(11) para el diagnóstico del SGB o con alguna de sus variantes(2,22,24). Aunque los estudios de velocidad de conducción y electromiografía pueden aportar al diagnóstico no se consideraron obligatorios, criterio que se ha aceptado en otras investigaciones similares(25).
Registro de datos
Se elaboró un formulario especial para esta investigación con el fin de registrar la información de las historias clínicas. Para completar la caracterización de los pacientes, se infirió el grado de paresia mediante la escala funcional de Hughes en el nadir de la enfermedad (0: normal; 1: signos o síntomas menores, capaz de correr; 2: puede caminar cinco metros sin ayuda, independientemente; 3: puede caminar cinco metros con un andador o soporte similar; 4: no puede caminar, permanece en cama o silla de ruedas; 5: requiere asistencia ventilatoria mecánica, AVM; 6: muerte) (6,8). Se registraron todas las otras hipótesis diagnósticas planteadas en las historias clínicas de los pacientes incluidos y los estudios realizados para descartarlas. Se registraron datos relevantes del tratamiento y del pronóstico inmediato. Se consideró que presentaban una disociación albumino-citológica en el LCR cuando la proteinorraquia era mayor de 0,45 g/L, y la celularidad de no más de 10 leucocitos (mononucleares)/mm3. Se incluyeron, con un análisis especial, aquellos con hasta 50 leucocitos(11).
Análisis estadístico
Las variables se expresaron en frecuencias absolutas y relativas. Las variables cuantitativas se expresaron mediante medidas de tendencia central (media y mediana) y medidas de dispersión (rango). Para las variables principales, se calculó el intervalo de confianza del 95% (IC 95%). Para el análisis de las diferencias de entre proporciones en las variables dicotómicas se utilizó la prueba no paramétrica de chi al cuadrado. Para los cálculos mencionados se utilizó el programa Epi-Info 6.
La población de 82 pacientes requirió, en total 1.721 días de hospitalización, con una media de 21,5 días y una mediana de 15 días (rango: 4–103 días). De los 82 pacientes, 59 (72%, IC95% 61%-81%) requirieron su ingreso en una unidad de cuidados intensivos pediátricos (UCIP). En total, los ingresos en las UCIP implicaron 855 días (49% del total de los días de hospitalización), con una media de 12 días y una mediana de 4 días (rango: 1-102 días). En 9 pacientes (10,9%), la duración de la estancia en la UCIP fue superior a un mes.
Antecedentes inmediatos
En las historias clínicas de 62 pacientes (76%, IC95%: 65-84%) se registró la existencia de un antecedente de vínculo infeccioso en los 30 días antes del inicio de las manifestaciones neurológicas: infección respiratoria aguda (54%), gastroenteritis (13%), asociación de infección respiratoria y gastroenteritis (6%), e inmunización con vacuna antimeningocócica B-C (2,4%).
Tiempo de evolución de las manifestaciones clínicas neurológicas en el ingreso en el CHPR
En 32 pacientes (39%), que habían sido referidos de otros hospitales, no fue posible determinar con certeza la fecha de ingreso en el hospital de origen. Los 50 pacientes que fueron hospitalizados directamente en el CHPR tenían un tiempo medio de evolución de su enfermedad de cuatro días. En 20 de los 50 pacientes (40%), la enfermedad tenía un día o menos de evolución y en 12 pacientes (24%), la enfermedad se había iniciado más de una semana antes del ingreso.
Síntomas neurológicos iniciales
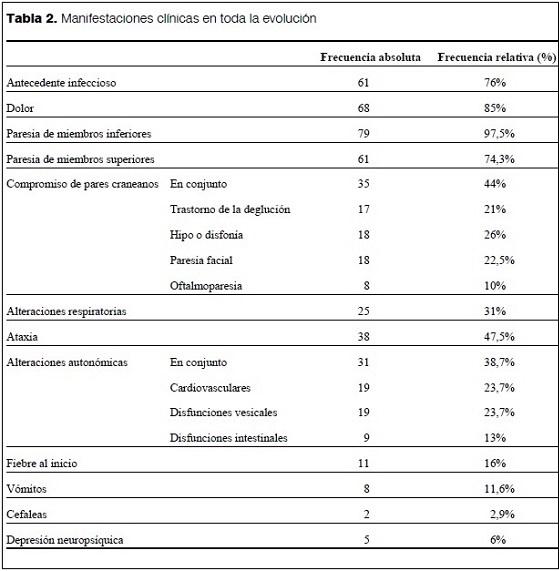
Los pacientes con SGB se presentaron inicialmente con diferentes tipos de síntomas neurológicos, como dolor, paresia de las extremidades, afectación de los pares craneales, elementos de ataxia o combinaciones de los diferentes tipos de síntomas (tabla 1).

Síntomas sensitivos
En las historias clínicas de 68 pacientes (83% IC95% 72-90%) se registró la presencia de dolor. Cuatro pacientes (5%) presentaron parestesias y 2 pacientes (2,5%) refirieron cefaleas en algún momento de la evolución.
La localización más frecuente del dolor fue en los miembros inferiores (58 pacientes, 71% del total). Ninguno presentó déficit sensitivo o nivel sensitivo definido (tablas 1 y 2).
Síntomas motores
De los 82 pacientes, 80 (97,5%) presentaron paresia de los miembros inferiores y 61 (74,3%) paresia de los miembros superiores. Los síntomas de la alteración de la función respiratoria, como tos débil, retención de secreciones o disminución de la capacidad vital, se presentaron en 21 (25,6%) pacientes.
Ataxia
Treinta y ocho pacientes (46% IC95% 35-57%) presentaron uno o más signos clínicos que se interpretaron como correspondientes a ataxia y que, analizados individualmente, incluyeron: inestabilidad en la sedestación en 7 (8,5%), aumento de la base de sustentación en 15 (18%), inestabilidad en la marcha en 27 (33%), dismetría en 3 (6%) y disartria en 4 (5%).
Manifestaciones clínicas autonómicas
Se registraron manifestaciones cardiovasculares en 23% de los pacientes (hipertensión arterial en 13, taquicardia en 3 y ambas alteraciones en otros 3), disfunciones vesicales en 23% de los pacientes (retención de orina e incontinencia urinaria) y disfunciones intestinales en el 11% de los pacientes (trastornos de la defecación y estreñimiento). Consideradas en su conjunto, las alteraciones autonómicas se registraron en 31 (39%) pacientes.
Otras manifestaciones clínicas no típicas del SGB
De los 82 pacientes incluidos, 11 (16%) presentaron fiebre al inicio de los síntomas neurológicos. Ocho pacientes (11,6%), presentaron vómitos y 5 (6%) depresión neuropsíquica.
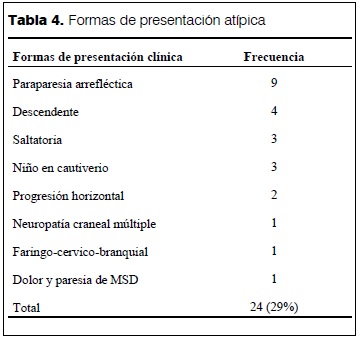
Formas de presentación atípica
De un total de 82 pacientes, 24 (29%) se presentaron de forma no habitual. En la tabla 4 se describen las distintas formas de presentación. A destacar son los 3 niños que presentaron la manifestación más severa de la enfermedad, como el síndrome del “niño en cautiverio” o “encerrado en sí mismo” (“locked-in syndrome”).
La puntuación de la escala de valoración funcional de Hughes en el peor momento de la evolución (nadir) fue de 2 en siete pacientes (8,7%), de 3 en trece pacientes (16%), de 4 en 43 pacientes (52,4%) y de 5 en 19 pacientes (23%).
Estudios del LCR
En 78 historias clínicas se registraron los resultados cuantitativos del estudio citoquímico del LCR de, por lo menos, una punción lumbar (PL). En 16 de esos pacientes, se realizaron dos punciones lumbares. En las historias clínicas restantes figuraba que el estudio del LCR era normal, pero no se transcribieron los valores de proteinorraquia y celularidad.
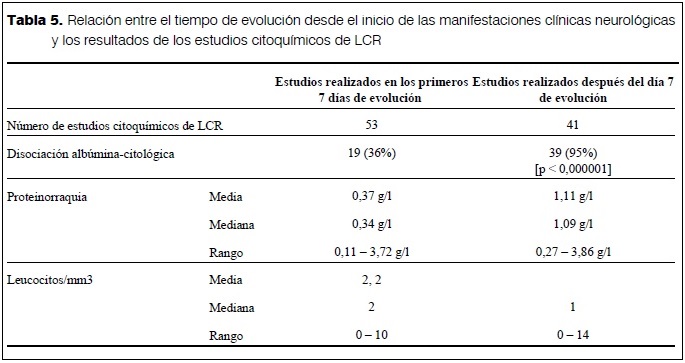
La primera PL se realizó entre el primer día y el día 30 de evolución de la enfermedad neurológica (media: 6,5 días, mediana: 5 días). Dado que la probabilidad de que la prueba sea positiva depende fundamentalmente del tiempo de evolución de las manifestaciones neurológicas, se compararon los resultados de las PL realizadas en los primeros siete días de evolución con los resultados de las PL realizadas posteriormente. Mientras que en los LCR obtenidos en la primera semana de evolución se observó disociación albúmino-citológica en 19/53 (36%), los LCR obtenidos de PL realizadas luego de la primera semana mostraron disociación albúmino-citológica en 39/41 (95%) (p < 0,000001) (tabla 5).
Los estudios virológicos realizados en 34 (41%) pacientes incluyeron el estudio para los virus de la familia herpes en todos los casos y el estudio para enterovirus en 12 de los 34 pacientes. Solo un paciente presentó PCR positiva para citomegalovirus. De los 34 pacientes, sólo en nueve casos se plantearon diagnósticos diferenciales con enfermedades neurológicas centrales infecciosas o posinfecciosas.
En 23 pacientes (28%) se realizó el estudio inmunoelectroforético del LCR. En 10 de ellos se informó de un aumento de la permeabilidad de la membrana hematoencefálica, tres mostraron una leve síntesis intratecal de inmunoglobulinas G y en ninguno se informó de la presencia de bandas oligoclonales locales.
En 44 historias clínicas (53%) se registró el resultado del estudio de velocidad de neuroconducción y electromiograma. El patrón neurofisiológico de la polineuropatía se informó como desmielinizante en 11 pacientes, axonal en 19 y mixto (axonal y desmielinizante en diferente proporción) en otros 12 casos. Estos estudios fueron realizados con una media de 13 días de iniciada la enfermedad (rango 3–160 días).
Diagnósticos diferenciales
En 35 historias clínicas (43%) se registraron entidades diagnósticas diferentes del SGB en algún momento de la evolución, que en conjunto sumaron 58 hipótesis diagnósticas. Se plantearon como diagnósticos diferenciales, de mayor a menor frecuencia: mielopatia aguda, encefalomielitis difusa aguda, encefalitis, miositis aguda inflamatoria, meningoencefalitis, intoxicación, ataxia cerebelosa, entre otros.
En total se realizaron 70 estudios paraclínicos para descartar diferentes diagnósticos diferenciales (tabla 6). A 35 pacientes (43%) se les practicaron, en total, 41 estudios neuroimagenológicos. Tres de estos pacientes corresponden a aquellos con síndrome de “niño encerrado en sí mismo” a quienes se le realizó resonancia de cráneo descartando de esta manera el compromiso pontino. En una de las resonancias magnéticas (RM) de médula se observó un realce meníngeo con la administración del contraste paramagnético en la médula terminal y las raíces de la cola de caballo.

Soporte ventilatorio
19 pacientes recibieron soporte ventilatorio con AVM (23%, IC95%: 13%–31%) de los cuales 11 requirieron posteriormente ventilación no invasiva (VNI). Otros dos pacientes recibieron solo VNI en toda la evolución de la enfermedad.
Inmunoterapia
En total 67 pacientes (82%, IC95%: 71%–89%) recibieron tratamiento con inmunoglobulinas intravenosas, tres de ellos (6%) fueron tratados además con plasmaféresis y once (13.4%) corticoterapia intravenosa.
Pronóstico inmediato
No hubo fallecidos en el egreso hospitalario (mortalidad: 0%; IC 95%: 0%–4%) y se otorgó el alta a todos los pacientes con la sintomatología en regresión.
Discusión
Los 82 pacientes pediátricos con SGB incluidos en este estudio representan un número importante para nuestro país, comparable a otros estudios similares realizados en otros países de la región.
El rango de edades fue muy amplio dado que abarcó entre 6 meses y 14 años y 9 meses, aunque predominaron los pacientes de menor edad. Al igual que en otras series, predominó el sexo masculino(7,26).
El diagnóstico temprano del SGB puede ser difícil. Aunque este síndrome se presenta generalmente como una enfermedad aguda, casi el 20% de los pacientes que ingresaron directamente en el CHPR instalaron sus síntomas en forma subaguda y presentaron más de una semana de evolución en el ingreso. En segundo lugar, los síntomas iniciales pueden ser muy variados e inespecíficos. El 51% presentó, como primer síntoma, dolor de forma aislada. Este predominó en los miembros inferiores, pero algunas veces fue unilateral y otras veces se asoció con dolor en otras regiones del organismo. Más difícil aún puede resultar el diagnóstico temprano en aquellos pacientes que comenzaron con una parálisis facial periférica unilateral (dos casos), con trastornos de la deglución o con ataxia como manifestación clínica aislada. En este estudio, el inicio con afectación de los pares craneales correspondió al 4% de los casos, valor similar al 5% mencionado en los criterios de Asbury y colaboradores(11).
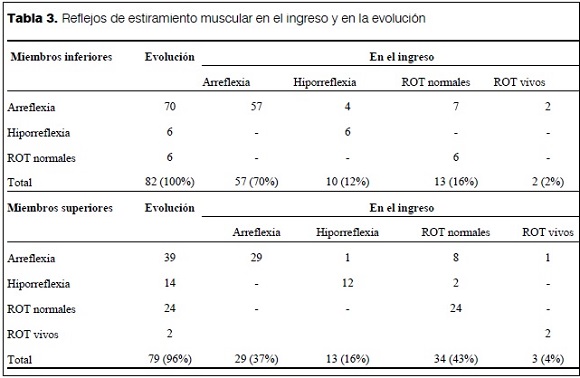
Si bien la forma de presentación clásicamente descrita es una parálisis ascendente arrefléctica, los reflejos de estiramiento muscular –en el momento del ingreso– eran normales o aumentados en los miembros inferiores en uno de cada seis pacientes y en los miembros superiores en casi la mitad de los pacientes. Aunque se describen como síntomas atípicos del SGB(11), 32% de los pacientes presentó, junto con las manifestaciones clínicas iniciales, fiebre (16%), cefaleas (3%), depresión neuropsíquica (6%) o vómitos (12%).
Otra forma de valorar las dificultades que plantea el diagnóstico temprano del SGB se revela a través de los diagnósticos diferenciales explicitados en el 41% de las historias clínicas. La naturaleza de las enfermedades planteadas incluyó patologías infecciosas, desmielinizantes postinfecciosas, tumorales, tóxicas, vasculares, musculares, osteoarticulares, psiquiátricas, etcétera.
Con el fin de descartar esas otras patologías, se realizaron en total 70 estudios complementarios que incluyeron 41 estudios imagenológicos del SNC a 35 pacientes (42,6%).
En relación con las características clínicas en la evolución, la paresia fue la manifestación clínica presente en el 100% de los pacientes de este estudio. El 97,5% presentó la paresia de los miembros inferiores, mientras que en un 74,3% se registró la paresia de los miembros superiores. El 44% de los pacientes presentó la afectación de los pares craneales, que, considerados por la función afectada, 21% presentó trastornos de la deglución, 26% hipofonía o disfonía, 22% parálisis facial periférica –unilateral en la tercera parte de los casos– y 10% oftalmoparesia extrínseca. Esto coincide, globalmente, con lo descrito en la literatura, que señala que el 20%–50% presenta debilidad facial, generalmente asimétrica, 25% afectación bulbar y 8%–10% paresia de oculomotores(6).
En nuestro estudio, 25% presentó síntomas relacionados con la alteración de la función respiratoria y el 23% requirió AVM, proporción similar a la descrita en la literatura(6).
El 83% de los pacientes (IC95% 72%–90%) refirió dolor, que en su gran mayoría se localizó en los miembros inferiores, asociado en casi la mitad de los casos a dolor en otras localizaciones.
En 46% de los pacientes (IC95%: 35%–57%) se registraron manifestaciones clínicas de ataxia. Este valor concuerda con el mencionado en la literatura, que indica que casi la mitad de los pacientes se presenta con “ataxia”(5).
Las variantes atípicas del SGB incluyeron nueve pacientes con paraparesia, tres casos con variantes saltatorias, cuatro se presentaron con una progresión descendente, dos con progresión horizontal, un paciente con neuropatía craneal múltiple, un paciente con dolor y paresia de miembro superior derecho y un paciente con una forma faringocervicobraquial con ataxia (elemento del SMF). En total, representaron 29% de la serie. Este valor es incluso mayor que el reportado por Buompadre y colaboradores(22) que publicaron una serie de 20 pacientes con variantes inusuales del SGB en la infancia, lo que representaba 11% de los pacientes con SGB internados en el Hospital Garrahan (Buenos Aires, Argentina) en 12 años (1993-2004). Tres pacientes de nuestra serie presentaron una forma rápidamente progresiva, llegando a una cuadriplejia flácida generalizada con pérdida absoluta de movimientos y reflejos, lo que constituye un cuadro clínico denominado “niño en cautiverio’, que se asemeja a la muerte cerebral(27).
El estudio citoquímico del LCR planteó –y seguirá planteando– un problema difícil de resolver. La mayoría de los pacientes ingresó en la primera semana de evolución de la enfermedad. Era necesario “confirmar” el diagnóstico y, en algunos casos, descartar diagnósticos diferenciales, para lo que se realizó una PL. Sin embargo, la disociación albúmino-citológica se observó en 36% de los estudios realizados en la primera semana de evolución. La situación cambió radicalmente después de esta primera semana, cuando 95% de los LCR mostraron la disociación albúmino-citológica. Habitualmente se menciona como un criterio diagnóstico de SGB el aumento de la concentración de proteínas en el LCR después de la primera semana de enfermedad, pero en la literatura revisada anteriormente no se habían encontrado referencias a qué ocurría cuando la PL se realiza en la primera semana. Este estudio permitió establecer que un valor negativo de dicha prueba no descarta el SGB cuando la PL se realiza en la primera semana y un valor positivo significativamente alto cuando la PL se realiza luego de la primera semana.
El pronóstico vital inmediato fue bueno y no se registraron fallecimientos en esta serie de casos (mortalidad: 0%; IC95% 0%–4%). Tampoco se registraron defunciones en una serie de 179 pacientes ingresados por SGB en el Hospital Garrahan (mortalidad: 0%; IC95% 0%–2%) (5). Sin embargo, en la serie de 17 pacientes pediátricos de un hospital de Portugal con SGB, ingresados entre 1994 y 2003, se registró una defunción (mortalidad: 5,9%; IC95% 0,3%–30%) (26).
Conclusiones
- Aunque el SGB es relativamente infrecuente, la revisión de las hospitalizaciones en el principal hospital pediátrico del país logró obtener información sobre un número importante de casos.
- Los pacientes pediátricos con SGB generalmente requieren hospitalizaciones prolongadas e ingresos en la UCIP para monitorización y/o tratamiento.
- Las manifestaciones clínicas del SGB pueden ser muy diversas, lo cual plantea dificultades importantes en la orientación clínica inicial. Es fundamental conocer las formas de presentación típica, las variantes y los diagnósticos diferenciales.
- En la primera semana de evolución, el estudio citoquímico del LCR para buscar la disociación albuminocitológica tiene una sensibilidad muy baja para apoyar el diagnóstico de SGB. Un resultado normal de las proteínas en el LCR no debe alejar el diagnóstico de SGB si el cuadro clínico sugiere éste o de alguna de sus variantes. En ese caso se deberá considerar la posibilidad de reiterar la PL después del séptimo día de evolución de la enfermedad o se podrá recurrir a otro tipo de exámenes paraclínicos, como los estudios electrofisiológicos, para confirmar dicho diagnóstico.
- A pesar de tratarse de una patología grave, con riesgo de mortalidad, en esta serie no se registraron fallecimientos, lo que coincide con otras casuísticas del SGB en pediatría.
Referencias bibliográficas
1. Sladky JT. Guillain-Barré syndrome in children. J Child Neurol 2004; 19(3):191-200.
2. Levin KH. Variants and mimics of Gullain-Barré syndrome. Neurologist 2004; 10(2):61-74.
3. Vajsar J, Fehlings D, Stephens D. Long-term outcome in children with Guillain- Barré syndrome. J Pediatr 2003; 142(3):305-9.
4. Van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol 2008; 7(10):939-50.
5. Sladky JT. Guillain-Barré syndrome. En: Jones HR, DeVivo DC, Darras BT, eds. Neuromuscular disorders of infancy childhood and adolescence – a clinician’s approach. Philadelphia: Elsevier Science, 2003:407-24.
6. Buompadre MC, Gañez LA, Arroyo HA. Síndrome de Guillain-Barré: espectro clínico y actualizaciones. Med Infant 2005; 12(3):207-13.
7. Hughes RAC, Cornblath DR. Guillain-Barré syndrome. Lancet 2005; 366(9497): 1653-66.
8. Kuwabara S. Guillain-Barré syndrome: epidemiology, pathophysiology and management. Drugs 2004; 64(6):597-610.
9. Tellería-Díaz A, Calzada-Sierra DJ. Síndrome de Guillain-Barré. Rev Neurol 2002; 34(10):966-76.
10. Van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Peripher Nerv Syst 2005; 10(2):113-27.
11. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain- Barré syndrome. Ann Neurol 1990; 27(Suppl):S21-4.
12. Uncini A, Kuwabara S. Electrodiagnostic criteria for Guillain–Barré syndrome: a critical revision and the need for an update. Clin Neurophysiol 2012; 123(8):1487–95.
13. Kokubun N, Nishibayash M, Uncini A, Odaka M, Hirata K, Yuki N. Conduction block in acute motor axonal neuropathy. Brain 2010; 133(10):2897-908.
14. Mogale KD, Anthony JH, Ryan MM. The pharyngeal-cervicalbrachial form of Guillain-Barré syndrome in childhood. Pediatr Neurol 2005; 33(4):285-8.
15. Chiò A, Cocito D, Leone M, Giordana T, Mora G, Mutani R, et al. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology 2003; 60(7):1146-50.
16. Badía Picazo MC, Santoja Llabata JM, Gil Gimeno R, Salvador Aliaga A, Pascual Lozano AM, Láñez-Andrés JM. Hiperreflexia en un paciente con síndrome de Guillain-Barré. Rev Neurol 2004; 38(7):697-8.
17. Polo JM, Alaña-García M, Cacauelo-Pérez P, Ortín-Castaño A, Ciudad-Bautista J, López-Alburquerque JT. Síndrome de Guillain-Barré atípico: neuropatía craneal múltiple. Rev Neurol 2002; 34(9):835-7.
18. Overell JR, Willison HJ. Recent developments in Miller Fisher syndrome and related disorders. Curr Opin Neurol 2005; 18(5):562-6.
19. Winer JB. Bickerstaff’s encephalitis and the Miller Fisher syndrome. J Neurol Neurosurg Psychiatry 2001; 71(4):433-5.
20. Okumura A, Ushida H, Maruyama K, Itomi K, Takahashi M, Osuga A, et al. Guillain-Barré syndrome associated with central nervous system lesions. Arch Dis Child 2002; 86(4):304-6.
21. Saad M, Youssef S, Kirschke D, Shubair M, Haddadin D, Myers J, et al. Acute flaccid paralysis: the spectrum of a newly recognized complication of West Nile virus infection. J Infection 2005; 51(2):120-7.
22. Buompadre MC, Gañez LA, Miranda M, Arroyo HA. Variantes inusuales del síndrome de Guillain-Barré en la infancia. Rev Neurol 2006; 42(2):85-90.
23. Cerisola A, Capote G, Scavone C. Síndrome de Guillain-Barré en pediatría: diferentes formas de presentación y dificultades en el diagnóstico precoz. Rev Neurol. 2007; 44(12):725-32.
24. Ropper AH. Further regional variants of acute immune polyneuropathy. Arch Neurol 1994; 51(7):671-5.
25. Korinthenberg R, Schessl J, Kirschner J, Shulte Mönting J. Intravenously administered immunoglobulin in the treatment of childhood Guillain-Barré syndrome: a randomized trial. Pediatrics 2005; 116(1):8-14.
26. Monteiro JP, Fonseca S, Proenca J, Calhau P, Braga M, Fonseca MJ. Síndrome de Guillain-Barré en edad pediátrica. Experiencia de la unidad de neuropediatría de un hospital portugués. Rev Neurol 2006; 42(3):144-9.
27. Medici C, Gonzalez G, Cerisola A, Scavone C. Locked in syndrome in three children with Guilain-Barré syndrome. Pediatr Neurol 2011; 44(1):61-4.
Correspondencia: Dra. Lucía Cibils.
Correo electrónico: lucibils@yahoo.com


{kind=link}