










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.84 no.2 Montevideo 2013
Hiperoxaluria primaria en tres hermanos
Margarita Halty1, Marina Caggiani2, Oscar Noboa3
1. Prof. Adj. Pediatría. Nefrólogo. Medicina Personalizada (MP).
2. Ex Prof. Adj. Pediatría. Nefrólogo. Medicina Personalizada (MP).
3. Prof. Nefrología. Medicina Personalizada (MP)
Fecha recibido: 31 de agosto de 2012.
Fecha aprobado: 4 de junio de 2013.
Resumen
La hiperoxaluria es responsable de 2%-20% de las causas metabólicas de litiasis en niños y adolescentes. Esta puede ser secundaria o primaria (HP), en este último caso es tipo I o tipo II, de herencia autosómica recesiva.
El objetivo es analizar diagnóstico y evolución en dos hermanos con HP tipo I y un tercero con alta probabilidad.
El primer caso presentó a los 9 años un cólico nefrítico con litiasis múltiples bilaterales y una IRA leve. Presentaba hiperoxaluria de 214 mg/1,73 m2. Se descartaron causas secundarias. El estudio genético demostró dos copias de la mutación IIe244Thr. Los otros dos pacientes se presentaron en forma similar, a edades de 8 y 17 años. La piridoxina a altas dosis descendió los niveles de oxaluria como está descrito en un tercio de los casos. El tiempo de evolución es 7, 3 y 1 año respectivamente. Persisten actualmente con litiasis bilaterales, sin nefrocalcinosis ni alteraciones del medio interno. La severidad de la HP tipo I es variable, hay formas de presentación temprana con litiasis recurrente y falla renal crónica en la infancia o en la adolescencia; otras de inicio tardío en edad adulta.
Se trata de una enfermedad grave, progresiva, cuyo diagnóstico temprano con estudio metabólico completo puede mejorar el pronóstico, fundamentalmente en aquellos que responden a la piridoxina. El manejo de la litiasis y sus complicaciones es fundamental para evitar la IRC. Cuando ésta se desarrolla, la diálisis agresiva y el trasplante hepatorrenal son las opciones terapéuticas. Es el primer informe de casos confirmados de hiperoxaluria primaria en nuestro país.
Palabras clave:
HIPEROXALURIA PRIMARIA
UROLITIASIS
Summary
Hyperoxaluria (HP) is responsible of 2%-20% of the metabolic causes of lithasis in children and adolescents. This can be secondary or primary. The primary one may be type I or II, its inheritance is autosomal recessive. The objective of this issue is to analyse the diagnosis and outcome of two siblings type I HP and a third one who probably also has it. At age 9 the first case presented a nephritic colic, multiple bilateral lithiasis and a mild renal failure. She had a hyperoxaluria of 214 mg/ 1,73 m2 sc. Secondary causes were discarded. The genetic test showed two copies of the Ile244Th gene mutation. The other two patients presented similar clinical features at 8 and 17 years old. Pyridoxine in high doses decreased oxaluria levels as it is described in one third cases. The patients' follow up were of 7, 3 and 1 year each. They persist with bilateral lithiasis, without nephrocalcinosis and no homeostasis alterations. The severity of HP type I is variable, some patients present at an early age recurrent lithiasis. Others suffer from severe renal failure in their childhood and adolescence, meanwhile others present symptoms in adultness.
HP type I is a progressive and severe illness. Its early diagnosis can improve its outcome mainly in those who respond to pyridoxine. The management of lithiasis and its complications is very important to avoid chronic renal failure. In this last situation aggressive dialysis, liver and kidney transplantation are therapeutic options. This is the first report of primary HP type I in our country.
Key words:
HYPEROXALURIA, PRIMARY
UROLITHIASIS
Introducción
Aunque la urolitiasis tiene una incidencia y prevalencia 50-75 veces menor en la edad pediátrica que en adultos, actualmente está siendo reconocida con mayor frecuencia. La hiperoxaluria es responsable de 2%–20% de las causas metabólicas de litiasis en niños y adolescentes y se manifiesta por enfermedad litiásica renal y/o nefrocalcinosis. Se define hiperoxaluria cuando la excreción urinaria en 24 horas supera el valor de 45 mg/1,73 m2 superficie corporal (SC) (1,2).
La hiperoxaluria puede resultar de un incremento en la absorción intestinal (secundaria o entérica) o de sobreproducción endógena: hiperoxaluria primaria (HP). Esta última se debe a un error congénito en el metabolismo del glioxilato hepático, siendo de herencia autosómica recesiva (1). Hay dos tipos de HP: en la HP tipo I existe un déficit funcional en la enzima alaninaglioxilato aminotransferasa (AGT) a nivel de los peroxisomas hepáticos, por lo que no se produce la transaminación de glioxilato en glicina, sino que se deriva a la oxidación en oxalato o la reducción a glicolato, acumulándose ambos metabolitos (3). En la HP tipo II el déficit ocurre en la enzima glioxilato reductasa/hidroxipiruvato reductasa. La HP tipo I es la forma más frecuente y severa (1).
El exceso de oxalato producido se excreta a nivel renal, llevando a una sobresaturación urinaria de oxalato de calcio que puede ocasionar litiasis, nefrocalcinosis e insuficiencia renal. La ERC es secundaria a los reiterados episodios de obstrucción e infección, a los depósitos de oxalato de calcio y a sus efectos tóxicos en túbulos renales e intersticio (1).
El objetivo de la presentación es analizar la presentación y evolución diferentes en tres hermanos con enfermedad litiásica severa, dos de ellos con HP tipo I y el tercero con alta probabilidad de la misma afección.
Pacientes
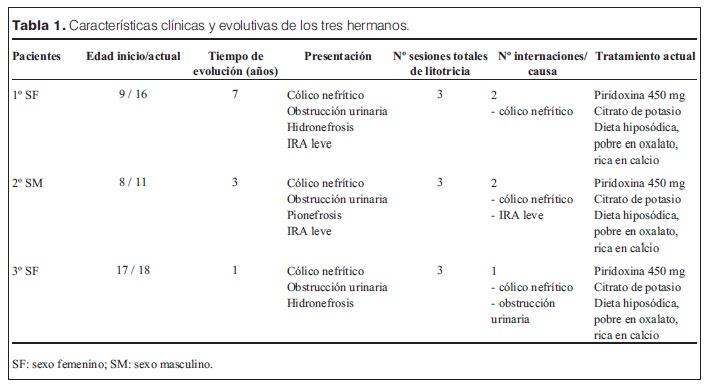
El primer caso es una paciente de sexo femenino que presentó a los 9 años un cólico nefrítico.
La ecografía y la TAC mostraron litiasis múltiples bilaterales.
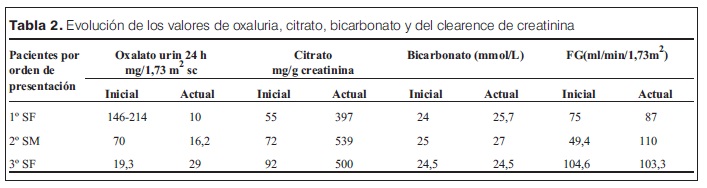
Del estudio de la litogénesis surge una hiperoxaluria con valores que alcanzaron 214 mg/1,73 m2 SC e hipocitraturia. Se descartaron causas secundarias de hiperoxaluria.
El estudio genético realizado en el Centro de Hiperoxaluarias de la Clínica Mayo demostró la presencia de dos copias de la mutación I244T.
Se inició tratamiento médico con altas dosis de piridoxina (450-500 mg/día), que tuvo buena respuesta, con descenso de la oxaluria a valores normales.
Actualmente tiene 16 años. La ecografía muestra riñones de tamaño conservado, con ecogenicidad discretamente aumentada, litiasis bilaterales múltiples, entre 2 y 6 mm de diámetro, sin dilatación de cavidades.
No se logró el estudio de sus hermanos en esa oportunidad.
El segundo caso es el de su hermano que presentó a los 8 años dolor abdominal intermitente durante 8 meses, diagnosticándose por ecografía litiasis bilaterales múltiples con ureterohidronefrosis bilateral y pionefrosis a derecha. Se realizó nefrostomía derecha y litotricia bilateral. Presentó una insuficiencia renal aguda (IRA) leve de causa obstructiva que retrocedió luego de la desobstrucción. El estudio químico de los fragmentos mostró oxalato de calcio. El estudio urinario demostró hiperoxaluria e hipocitraturia. No reiteró episodios obstructivos luego de iniciado el tratamiento médico.
Actualmente tiene 11 años, la ecografía muestra riñones de tamaño normal, ecogenicidad discretamente superior a la normal con buena diferenciación corticomedular. Presenta tres litiasis, dos a derecha de 2 mm y una a izquierda de 3,5 mm, sin dilatación de cavidades.
El tercer caso corresponde a la hermana mayor, que debutó a los 17 años con dolor lumbar de tipo cólico por lo que se realizó una ecografía diagnosticándose tres litiasis a derecha, de entre 11 y 13 mm, sin dilatación de cavidades. En un segundo episodio presentó obstrucción a nivel de la unión pieloureteral e hidronefrosis. Se realizó litotricia concomitantemente con el inicio del tratamiento médico. No se comprobó hiperoxaluria antes del inicio del tratamiento médico pero la composición química de los fragmentos de litiasis eliminadas postlitotricia fue oxalato de calcio. La citraturia fue baja y la oxaluria normal en una sola oportunidad previo al inicio del tratamiento.
Actualmente tiene 18 años, la ecografía muestra riñones de tamaño y ecogenicidad conservado, con dos litiasis de 3 mm a derecha y una de 8,5 mm a izquierda.
Los tres pacientes tuvieron siempre valores de calciuria y uricosuria normales.
Ninguno de ellos presentó nefrocalcinosis en el estudio tomográfico inicial, que no se repitió.
Se presentan dos tablas con una síntesis de la presentación clínica, tratamiento y evolución.
La familia autorizó a realizar esta comunicación.
Discusión
Se presentan tres casos de litiasis familiar con hiperoxaluria primaria confirmada en dos casos y de alta probabilidad en el tercero. La hiperoxaluria primaria tipo I tiene en Francia una incidencia de 1/120.000 recién nacidos vivos (4). Su severidad es variable, con irregular correlación con el fenotipo. Se describen cinco presentaciones: 1) forma infantil con nefrocalcinosis temprana y falla renal; 2) litiasis recurrente y falla renal progresiva durante la niñez o adolescencia; 3) inicio tardío con litiasis en la edad adulta; 4) diagnóstico postrasplante, con recurrencia de la litiasis; 5) diagnóstico en etapa presintomática, debido a una historia familiar (5).
La mutación hallada (Ile244Thr) y la mutación Gly170Arg son las dos más frecuentes en EE.UU. y Europa (6). Entre los pacientes homocigóticos para Ile244Thr, Santana y colaboradores constataron una gran variabilidad en la presentación clínica, con casos de insuficiencia renal en edades tempranas y otros que mantienen la función renal en la edad adulta. Esta falta de correlación genotipo-fenotipo es interpretada por el efecto de genes modificadores y/o factores ambientales que promueven o inhiben la cristalización de oxalato de calcio (7). La presentación clínica tuvo diferente severidad en la presentación y evolución de estos tres hermanos.
Tratándose de una enfermedad con herencia autosómica recesiva, con una probabilidad de 25% en cada hijo de presentar la enfermedad, llama la atención que dos se encuentren afectados y el tercero muy probablemente también. Sería importante realizar el estudio genético de los hermanos, fundamentalmente del tercer caso
En relación al tratamiento, deben realizarse esfuerzos intensivos para minimizar la cristalización de oxalato de calcio y la formación de cálculos en los riñones, lo que parece preservar la función renal (8). Aproximadamente un tercio de los pacientes con HP tipo I responden a dosis elevadas de piridoxina con una marcada reducción, y a veces normalización de la excreción urinaria de oxalato. Se desconoce exactamente su mecanismo de acción. La piridoxina es un cofactor de la AGT y podría favorecer el plegamiento correcto de algunas de las formas mutadas de la enzima (7). Gürgöze y colaboradores observaron que en los pacientes con hiperoxaluria los cálculos aumentan de tamaño a pesar del tratamiento médico bien conducido (9). También es útil el tratamiento con citrato y debería reducirse la ingesta de oxalato. Recientemente ha despertado interés la administración oral de bacterias como el Oxalobacter formigenes o de enzimas que degradan el oxalato en la luz intestinal. También estimulan la secreción de oxalato a nivel entérico (1,8).
El manejo de la litiasis es un desafío para el seguimiento a largo plazo de estos pacientes. Las complicaciones de la litiasis son la obstrucción y la infección, pudiendo evolucionar a la insuficiencia renal crónica. Los tres pacientes relatados presentaron la complicación obstructiva; la litotricia extracorpórea fue una opción válida para lograr la desobstrucción. En uno de ellos fue imprescindible la nefrostomía para desobstruir y tratar exitosamente la infección.
En otros casos, la ureteroscopía y la colocación de stent pueden ser útiles (6). Dos de los tres pacientes presentaron una insuficiencia renal aguda leve con evolución favorable.
La paciente 1 tiene un clearence de creatinina en valor límite, es portadora de una enfermedad renal crónica estadio 2. Los pacientes 1 y 2 presentan un aumento en la ecogenicidad, sin signos ecográficos ni tomográficos de nefrocalcinosis, otra posible evolución/ complicación de la enfermedad.
La bibliografía refiere que cuando el filtrado glomerular desciende por debajo de 30-50 ml/min/1,73 m2, la sobreproducción de oxalato con disminución de la excreción renal determina su depósito fundamentalmente a nivel óseo. Se producen depósitos vasculares de oxalato de calcio con compromiso de varios órganos y tejidos como: corazón, nervios, articulaciones, partes blandas, retina y otros (6).
En pacientes que desarrollan IRC estadío 4-5, la diálisis agresiva, seguida de trasplante hepático y renal, constituye el manejo más adecuado (8). En aquellos pacientes sin respuesta al tratamiento con piridoxina puede proponerse la realización de un trasplante preventivo (6).
La mitad de los pacientes alcanzan la IRC en etapa de sustitución de la función a los 25 años (6). El estudio oportuno de los hermanos en la etapa presintomática hubiera evitado parcialmente la formación de cálculos y sus consecuencias. A partir del diagnóstico, deben extremarse los esfuerzos para evitar elevaciones de la oxaluria y su precipitación mediante un tratamiento médico adecuado.
Se trata de la primera comunicación de casos de hiperoxaluria primaria en niños en nuestro país.
Referencias bibliográficas
1. Milliner DS. Urolithiasis. En: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, eds. Pediatric Nephrology. 6 ed. Boston: Springer, 2009:1405-30.
2. Hoppe B, Leumann E, Milliner D. Urolithiasis and nephrocalcinosis in childhood. En: Geary DF, Schaefer F, eds. Comprehensive Pediatric Nephrology. Philadelphia: Mosby, Elsevier, 2008:499-525.
3. Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol 2001; 12(9):1986-93.
4. Cochat P, Basmaison O. Current approaches to the management of primary hyperoxaluria. Arch Dis Child 2000; 82(6):470-3.
5. Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type I: still challenging! Pediatr Nephrol 2006; 21(8):1075-81.
6. Cochat P, Fargue S, Harambat J. Primary Hyperoxaluria. En: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, eds. Pediatric Nephrology. 6 ed. Boston: Springer, 2009:1069-79.
7. Santana A, Torres A, Salido E. Patología molecular de la hiperoxaluria primaria. Nefrología 2003; 23(supl. 1):90-7.
8. Alon US. Medical treatment of pediatric urolithiasis. Pediatr Nephrol 2009; 24(11):2129-35.
9. Gürgöze MK, Sarý MY. Results of medical treatment and metabolic risk factors in children with urolithiasis. Pediatr Nephrol 2011; 26(6):933–7.
Correspondencia: Dra. Margarita Halty. Correo electrónico: margahalty@hotmail.com


{kind=link}
{kind=link}