










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.82 no.4 Montevideo dic. 2011
Primer trasplante de progenitores hematopoyéticos en un paciente con inmunodeficiencia primaria en Uruguay: síndrome de hiper IgM ligado al X
Gustavo Dufort y Álvarez 1,2, Virginia Patiño 3, Carola López 4, Gabriela Sequeira 5,
Mariela Castiglioni 2, Carolina Pagés 2, Magdalena Schelotto 3, Elena Picheli 6, Jorge Decaro 7, Luis Castillo 8
1. Pediatra Hematooncólogo. Hospital Central de las Fuerzas Armadas.
2. Staff de la Unidad de Trasplante de Médula Ósea Pediátrica. Asociación Española.
3. Pediatra. Unidad Docente Asistencial de Inmunología e Infectología. Centro Hospitalario Pereira Rossell.
4. Pediatra Gastroenteróloga. Hospital Central de las Fuerzas Armadas.
5. Residente de Pediatría. Hospital Central de las Fuerzas Armadas.
6. Jefe del Departamento de Pediatría. Hospital Central de las Fuerzas Armadas.
7. Director Departamento de Medicina Transfusional. Asociación Española.
8. Director Unidad de Trasplante de Médula Ósea Pediátrica. Asociación Española.
Unidad de Trasplante de Médula Ósea Pediátrica. Asociación Española Primera de Socorros Mutuos.
Fecha recibido: 20 de setiembre de 2011.
Fecha aprobado: 15 de diciembre de 2011.
Resumen
Introducción: el trasplante alogénico de progenitores hematopoyéticos (TPH) es el único tratamiento de las inmunodeficiencias congénitas severas con potencial curativo. El síndrome hiper-IgM ligado al X tipo 1 (HIGM-1) -causado por mutaciones en el gen que codifica el ligando CD40- es una inmunodeficiencia primaria (IDP) con alta morbimortalidad a pesar de tratamientos de soporte adecuados, debido a infecciones recurrentes y colangiopatía. Describimos el primer TPH en una IDP en nuestro país en un niño con HIGM-1 con enfermedad hepática ya establecida.
Métodos: un paciente con HIGM-1 con infecciones reiteradas, falla de crecimiento, y colangiopatía documentada por biopsia hepática fue sometido a TPH alogénico de su hermana HLA idéntica. El condicionamiento fue no mieloablativo con fludarabina, melfalan y globulina antitimocítica.
Resultados: el niño lleva dos años del TPH. El quimerismo es 100% del donante. Está libre de infecciones, independiente de inmunoglobulina intravenosa y su colangiopatía impresiona resuelta. Los estudios de laboratorio confirman la corrección del defecto inmunológico.
Conclusión: el TPH alogénico de donante HLA idéntico realizado con condicionamiento no mieloablativo permitió la reconstitución inmune en un niño con HIGM-1 y colangiopatía preexistente, sin toxicidad hepática severa.
Palabras clave:
TRASPLANTE DE CÉLULAS MADRE
HEMATOPOYÉTICAS
SÍNDROMES DE INMUNODEFICIENCIA
LIGANDO CD40
CROMOSOMA X
ENFERMEDADES DE LOS CONDUCTOS
BILIARES
Summary
Introduction: allogenic stem cell transplantation (HSCT) has the potential to cure severe congenital immunodeficiencies. Hyper-IgM type 1 syndrome (HIGM-1) - caused by mutations in the gene encoding CD40 ligand – is a primary immunodeficiency with high morbidity and mortality despite adequate supportive care. We describe the first HSCT in our country in a child with HIGM-1 syndrome and preexisting cholangiopathy.
Methods: one child with HIGM-1 syndrome and persistent infections, growth failure, and cholangiopathy documented by liver biopsy underwent nonmyeloablative HSCT from HLA matched sibling with fludarabine, melphalan, and anti-thymocyte globulin as preparative regimen.
Results: the child is 2 years after his HSCT. Donor chimerism is 100%. He is free of infections and no longer depends on intravenous gammaglobulin. His cholangiopathy impresses resolved and laboratory assays shows correction of his immunologic defect.
Conclusion: nonmyeloablative HSCT from HLA matched sibling allowed immune reconstitution in a patient with HIGM-1 syndrome and preexisting cholangiopathy, without severe hepatotoxicity.
Key words:
HEMATOPOIETIC STEM CELL
TRANSPLANTATION
IMMUNOLOGIC DEFICIENCY SYNDROMES
CD40 LIGAND
X CHROMOSOME
BILE DUCT DISEASES
Introducción
Las inmunodeficiencias primarias (IDP) representan un grupo de enfermedades genéticamente heterogéneas que afectan distintos componentes del sistema inmune innato y adaptativo, como son los neutrófilos, macrófagos, células dendríticas, proteínas del complemento, células NK, y linfocitos T y B (1). La gran mayoría (90%) de estas deficiencias se presentan en edades pediátricas, especialmente antes de los primeros cinco años de vida, y se acompañan, a menudo, de infecciones que amenazan la vida. El trasplante de progenitores hematopoyéticos (TPH) puede ser el tratamiento de elección para curar la mayoría de las formas letales de IDP.
El síndrome de hiper IgM ligado al X (HIGM-1) es una inmunodeficiencia genética rara de los linfocitos T, causada por mutaciones en el gen que codifica el ligando CD40 (CD40L, CD154), glicoproteína normalmente expresada en la superficie de los linfocitos T activados (2,3), esencial para el cambio de isotipo de IgM a IgG, IgA e IgE, formación de los centros germinales y activación de los monocitos/macrófagos (4). Por lo tanto los pacientes con el síndrome HIGM-1 tienen niveles muy elevados o normales de IgM con marcada deficiencia de IgG, IgA e IgE, y ausencia de centros germinales en sus ganglios linfáticos. Estos pacientes tienen una inmunidad celular defectuosa que los hace susceptibles a infecciones oportunistas y frecuentemente desarrollan neutropenia (5).
Las manifestaciones clínicas incluyen infecciones respiratorias recurrentes, así como neumonía debido a infección con Pneumocystis jirovecii. Susceptibilidad a infección gastrointestinal con protozoos como Cryptosporidium parvum puede conducir a la colangitis esclerosante, cirrosis y colangiocarcinoma (6).
El tratamiento de soporte de estos pacientes incluye inmunoglobulina intravenosa (IGIV), profilaxis antibiótica contra infecciones oportunistas y factor estimulante de colonias de granulocitos (G-CSF) para la neutropenia (7).
Una significativa proporción de estos niños afectados fallecen tempranamente a pesar de tratamientos de soporte adecuados. El TPH es el único tratamiento que tiene el potencial de curar esta inmunodeficiencia de los linfocitos T.
Describimos el primer caso de una IDP, HIGM-1, tratado con TPH alogénico de hermano HLA idéntico en nuestro país.
Observación clínica
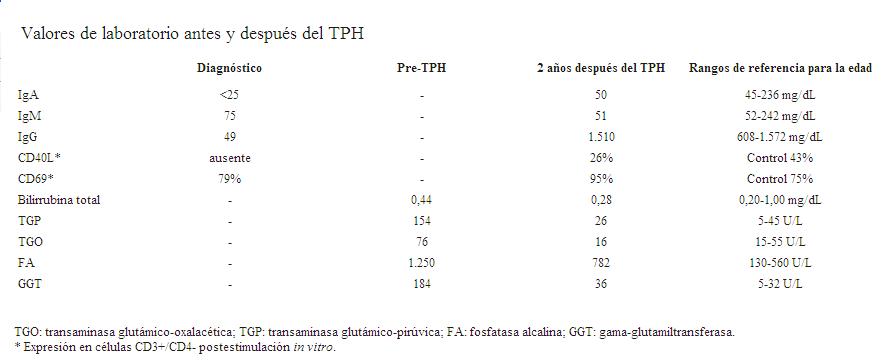
Nuestro paciente tenía como antecedente un hermano varón fallecido al año de vida en el curso de una infección respiratoria. A los 12 meses presentó su primera infección, una neumonía, sin aislamiento de germen. A los 15 meses, internación por desnutrición y diarrea, y a los 18 meses sepsis a Pseudomona aeruginosa por lo que estuvo 2 meses en unidad de cuidados intensivos. En esa oportunidad se estudió su inmunidad constatándose hipogamaglobulinemia severa con linfocitos B y T normales (tabla 1). Fue tratado con IGIV mensual y cotrimoxazol. A los 4 años presentaba falla de crecimiento y diarrea crónica. La biopsia de intestino muestra giardasis y quistes de Cryptosporidium. En ese momento se confirma el diagnóstico de HIGM-1 mediante la demostración de ausencia de expresión de CD40L en los linfocitos T activados. A los 5 años las enzimas hepáticas comienzan a elevarse y presenta neutropenia crónica que requiere tratamiento con G-CSF. La ecografía mostraba dilatación de la vía biliar intrahepática. Se realizó biopsia hepática que constató: parénquima hepático con arquitectura conservada; leve fibrosis periportal; proliferación ductular y fibrosis concéntrica alrededor de ductos biliares (catáfilas de cebolla); escaso infiltrado inflamatorio en los espacios porta; a nivel lobulillar hepatocitos conservados. Los hallazgos histológicos eran compatibles con el diagnóstico de colangitis esclerosante. Recibió tratamiento con ácido ursodeoxicólico sin una mejoría evidente. Debido al alto riesgo de progresión a la cirrosis y fallo hepático, la severa falla de crecimiento, la infección respiratoria recurrente y la disponibilidad de una hermana HLA-idéntica, a los 6 años se realizó el TPH. Para reducir la hepatotoxicicidad, el paciente recibió condicionamiento no mieloablativo (fludarabina 30 mg/m2/día por 5 días, melfalan 140 mg/m2 por un día, y gamaglobulina antitimocítica de conejo 2,5 mg/kg/día por 5 días). La profilaxis de la enfermedad injerto versus huésped (EICH) se realizó con ciclosporina A. Se realizó profilaxis infecciosa con cotrimoxazol, fluconazol, aciclovir, azitromicina y nitazoxanida. El paciente recibió progenitores hematopoyéticos obtenidos de sangre periférica (7,41 x 106 CD34+/kg) de su hermana de 1 año previamente estimulada con G-CSF. La aféresis requirió la colocación previa, bajo anestesia general, de dos vías venosas centrales en ambas venas femorales. Se utilizaron glóbulos rojos filtrados e irradiados para purgar el sistema del separador celular. El procedimiento no presentó complicaciones. A los 13 días el recuento absoluto de neutrófilos fue mayor de 1.000 elementos/mm3, y a los 30 días del trasplante el recuento de plaquetas fue mayor de 50.000 elementos/mm3. El día 28 se confirma un quimerismo 100% del donante. Previo al implante el paciente presentó alta tasa de diarrea, y hepatomegalia con aumento de la colestasis con cifras de bilirrubina directa de 11 mg/dl, fosfatasa alcalina de 5.500 UI/l y gama-glutamil transferasa de 1.800 UI/l. Presentó EICH grado II (piel +++) por lo que recibió tratamiento con corticoides sistémicos con buena evolución, manteniéndose la ciclosporina durante 90 días. La IGIV fue discontinuada a los 14 meses postrasplante. Los niveles de IgG e IgA se normalizaron y el análisis por citometría de flujo mostró expresión de CD40L en linfocitos T activados in vitro. Clínicamente el paciente mantiene un progresivo aumento de peso y talla, sin mayores infecciones y con un status funcional excelente. El hepatograma se normalizó, el hígado es de tamaño normal y no se encuentra evidencia ecográfica de dilatación de la vía biliar intra o extrahepática.
Discusión
Los primeros TPH realizados con éxito en humanos fueron reportados en 1968 en tres pacientes con IDP que recibieron trasplantes de hermanos HLA-compatibles. En los últimos 40 años ha habido importantes progresos en el TPH para IDP lo que ha permitido que la gran mayoría de los niños con IDP en el mundo desarrollado tengan acceso a donantes adecuados y procedimientos seguros que permiten corregir estas enfermedades de otro modo letales. Los mayores obstáculos para el éxito en la calidad de sobrevida y reconstitución inmune a largo plazo de los pacientes con IDP son: 1) retraso en el diagnóstico y 2) tratamiento en centros sin experiencia en el manejo de estos pacientes (8). Retraso en el diagnóstico de IDP o retraso en proceder al TPH, resulta frecuentemente en la emergencia de morbilidades pretrasplante, incluyendo infecciones oportunistas, exposición a virus herpes y desarrollo de complicaciones autoinmunes, todas las cuales pueden comprometer la probabilidad de éxito a largo plazo del TPH.
En Uruguay se realizan TPH en niños desde el año 1997 (9). Cerca de 250 trasplantes han sido realizados a la fecha que incluyen TPH autólogos y TPH alogénicos de donantes relacionados, no relacionados y donantes haploidénticos (10). En este periodo de tiempo entre 7 y 10 niños con Inmunodeficiencia Combinada Severa (IDCS) deberían haber sido diagnosticados en nuestro país. La IDCS representa la forma más grave de IDP, ya que el 100% de los casos tiene un curso fatal de no mediar una reconstitución inmune a través de un TPH alogénico. Su frecuencia ha sido estimada en 1 cada 75.000 a 100.000 nacimientos. Sin embargo hasta el momento ningún niño con IDCS ha recibido un TPH en nuestro país. Probablemente ha habido un subdiagnóstico de esta patología, o si fueron diagnosticados no han sobrevivido para tener acceso a un TPH.
El síndrome HIGM-1 se considera una IDCS (11), y comparte en muchos casos la severidad de este grupo de IDP, requiriendo en algunos casos un manejo y tratamiento similar. El TPH ha sido propuesto y aplicado con éxito en pacientes con síndrome HIGM-1 (12). La experiencia europea sugiere que 40%-50% de los pacientes pueden tener un curso benigno y sobrevivir más de 20 años con tratamiento de soporte adecuado. Sin embargo muchos pacientes desarrollan morbilidades crónicas en la primera década, incluyendo neutropenia, daño pulmonar y disfunción hepática (12). El daño pulmonar previo aparece como el factor de riesgo adverso más importante para el fracaso del TPH. Los pacientes con enfermedad hepática establecida, como es el caso de nuestro paciente, son un desafío a la hora de la planificación del tratamiento más adecuado. Es muy probable la reactivación de la infección oculta por Cryptosporidium en el postrasplante inmediato a pesar del uso de antimicrobianos profilácticos, y esto determinar un mayor compromiso de la función hepática. Regímenes de condicionamiento con fludarabina/melfalan causan menos toxicidad aguda que los regímenes convencionales y son preferibles en pacientes con enfermedad hepática preexistente (13). Nuestro paciente presentó tempranamente complicaciones vinculadas a la toxicidad del procedimiento y/o reactivación del Cyiptosporidium cuya evolución fue probablemente limitada por la recuperación temprana de los linfocitos T. La mejoría clínica y paraclínica de la enfermedad hepatobiliar en nuestro paciente es de particular interés porque es el reflejo de la reconstitución inmune con el consiguiente control del proceso infeccioso subyacente luego de un trasplante exitoso (14).
Es importante resaltar que este paciente podría haberse trasplantado antes, y probablemente en una situación clínica mejor si se hubiera recolectado la sangre de cordón umbilical (SCU) de su hermana y utilizado ésta como fuente de progenitores hematopoyéticos. Debería ser una práctica común la criopreservación de SCU de hermanos de pacientes con enfermedades pasibles de ser curadas con TPH.
Conclusiones
El TPH alogénico es un procedimiento riesgoso, especialmente en el contexto de pacientes con síndromes de inmunodeficiencia e infección. Los riesgos y beneficios del TPH en el HIGM-1 deben ser sopesados para decidir cuándo proceder al trasplante. Idealmente, los pacientes con un hermano compatible deberían ser considerados para TPH frente a los primeros signos de disfunción hepática. En el caso de nuestro paciente el trasplante fue realizado con enfermedad hepatobiliar ya establecida lográndose su mejoría tras el procedimiento como ya ha sido descrito (14). Otro factor que puede haber contribuido a la buena evolución es el régimen preparativo de intensidad reducida utilizado (13).
Referencias bibliográficas
1. Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, et al. Primary immunodeficiency diseases: An update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol 2007; 120(4): 776-94.
2. Korthauer U, Graf D, Mages HW, Brière F, Padayachee M, Malcolm S, et al. Defective expression of T-cell CD40 Ligand causes X-Linked immunode?ciency with hyper-IgM. Nature 1993; 361: 539-41.
3. Disanto JP, Bonnefoy JY, Gauchat JF, Fischer A, De Saint Basile G. CD40 Ligand mutations in X-linked immunodeficiency with hyper-IgM. Nature 1993; 361: 541-3.
4. Van Kooten C, Banchereau J. Functions of CD40 on B cells, dendritic cells and other cells. Current Opin Immunol 1997; 9: 330-7.
5. Andrews JF, Katz F, Jones A, Smith S, Finn A. CD40 Ligand deficiency presenting as unresponsive neutropenia. Arch Dis Child 1996; 74: 458-9.
6. Heyward AR, Levy J, Facchetti F, Notarangelo L, Ochs HD, Etzioni A, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol 1997; 158: 977-83.
7. Wang WC, Cordoba J, Infante AJ, Conley ME. Successful treatment of neutropenia in the hyper-immunoglobulin M syndrome with granulocyte colony-stimulating factor. Am J Pediatr Hematol Oncol 1994; 16: 160–3.
8. Filipovich AH. Hematopoietic cell transplantation for correction of primary immunodeficiencies. Bone Marrow Transplant 2008; 42: S49-S52.
9. Dufort y Alvarez G, Castiglioni M, Pagés C, Dabezies A, Decaro J, Castillo L. Trasplante de progenitores hematopoyéticos en pediatría: 10 años de experiencia. Arch Pediatr Urug 2008; 79(3): 201-9.
10. Dufort G, Pisano S, Incoronato A, Castiglioni M, Pages C, Simon E, et al. Feasibility and outcome of haploidentical SCT in pediatric high-risk hematologic malignancies and Fanconi anemia in Uruguay. Bone Marrow Transplant 2011. Obtenido de: www.ncbi.nlm.nih.gov/pubmed/21765479 [consulta: 20 ag. 2011].
11. Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol 2009; 124(6): 1161-78.
12. Gennery AR, Khawaja K, Veys P, Bredius RG, Notarangelo LD, Mazzolar E, et al. Treatment of CD40 ligand deficiency by hematopoietic stem cell transplantation: a survey of the European experience, 1993-2002. Blood 2004; 103: 1152-7.
13. Amrolla P, Gaspar HB, Hassan A, Webb D, Jones A, Sturt N, et al. Nonmyeloablative stem cell transplantation for congenital immunodeficiencies. Blood 2000; 96: 1239-46.
14. Jacobsohn DA, Emerick KM, Scholl P, Melin-Aldana H, O´Gorman M, Duerst R, et al. Nonmyeloablative Hematopoietic Stem Cell Transplant for X-Linked Hyper- Immunoglobulin M Syndrome With Cholangiopathy. Pediatrics 2004; e122-e127.
Correo electrónico: gdufort@chasque.net


{kind=link}