










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.81 no.3 Montevideo Sept. 2010
CASO CLÍNICO
Arch Pediatr Urug 2010; 81(3)
Síndrome de Alagille.
A propósito de un caso
Dras. Elsa Arocena 1, Karina Machado 2, María Catalina Pirez 3, Alicia Montano 4
1. Residente de Pediatría. Centro Hospitalario Pereira Rossell.
2. Profesora Adjunta de Clínica Pediátrica de la Facultad de Medicina.
3. Profesora Directora de la Clínica Pediátrica “A”, Facultad de Medicina.
4. Profesora Directora de la Clínica Pediátrica “B”, Facultad de Medicina.
Fecha recibido: 14 de junio de 2010.
Fecha aprobado: 9 de noviembre de 2010.
Resumen
El síndrome de Alagille es un trastorno autosómico dominante, con expresividad variable, cuya prevalencia es de 1 en 100.000 nacidos vivos. Se caracteriza por manifestaciones hepáticas, anomalías vertebrales y oculares, cardiopatía congénita y dismorfias faciales. Se ha identificado el defecto genético causante en el gen JAG 1, localizado en el cromosoma 20, del cual se describen diferentes mutaciones.
El pronóstico de este síndrome es variable y depende fundamentalmente de la entidad de la afectación hepática y los defectos cardiovasculares.
Se presenta un paciente de 10 años en el que durante una internación por una patología infecciosa adquirida, se realiza diagnóstico de síndrome de Alagille. Este paciente presenta cuatro de los cinco criterios mayores diagnósticos de la enfermedad: cardiopatía congénita, anomalías vertebrales, embriotoxon posterior y fascies característica. Además antecedentes familiares de cardiopatía congénita y dismorfias faciales, con una genealogía que recuerda el tipo de herencia autosómica dominante.
Palabras clave:
SÍNDROME DE ALAGILLE
ANOMALÍAS MÚLTIPLES
CARDIOPATÍAS CONGÉNITAS
OFTALMOPATÍAS
FACIES
HEPATOPATÍAS
Summary
Alagille Syndrome is an autosomal dominant disorder of variable expression, whose prevalence is 1 in 100 000 births, characterized by cholestasis, vertebral and eye abnormalities, congenital heart disease and characteristic facies. It is caused by mutations in the Jagged 1 gene. Several mutations have been described in this gene, located in chromosome 20. The prognosis of this syndrome is variable and depends on the severity of hepatic or cardiac involvement.
We present a 10 year old patient in which Alagille syndrome is diagnosed during a hospital stay for an infectious pathology. This patient has 4 of the 5 major features: characteristic facies, posterior embriotoxon, vertebral defects, congenital heart disease. Presents family history of congenital heart disease and facial dysmorphias with a genealogy that resembles an autosomal dominant disorder.
Key words:
ALAGILLE SYNDROME
ABNORMALITIES, MULTIPLE
HEART DEFECTS, CONGENITAL
EYE DISEASES
FACIES
LIVER DISEASES
Introducción
El síndrome de Alagille es un trastorno genético, autosómico dominante, que tiene un amplio espectro de presentaciones clínicas, que pueden ser variables, incluso en una misma familia (1-4). Se caracteriza por anomalías hepáticas, esqueléticas, renales, oculares, cardiovasculares y dismorfias faciales (1-7).
Fue descrito inicialmente en 1973 por Watson y Millar (6), pero fue en 1975 cuando Alagille describió los cambios histológicos y la asociación con otras manifestaciones clínicas (1). En 1987 en un informe de 80 casos Alagille y colaboradores sugirieron los criterios diagnósticos del síndrome: escasez de ductos intrahepáticos, patología cardiovascular, defectos vertebrales, embriotoxon posterior y fascies característica (2). La alteración genética fue localizada en el cromosoma 20 por Schnittger en 1989 (8).
Se ha citado como una de las causas más frecuentes de colestasis neonatal luego de la atresia biliar extrahepática y el déficit de alfa 1 antitripsina (9).
La prevalencia es de aproximadamente 1/100.000 nacidos vivos (10). En nuestro medio se publicó un caso, en 1994, de un paciente de 21 meses portador del síndrome en su forma completa con colestasis neonatal que evolucionó a insuficiencia hepatocítica e hipertensión portal (12). Se describen otros dos casos en una serie de 51 pacientes con colestasis neonatal en seguimiento en el servicio de gastroenterología del Centro Hospitalario Pereira Rossell entre los años 1983 y 2000 (13).
La causa del síndrome de Alagille es un defecto en el cromosoma 20 p12, donde se localiza el gen JAG 1, que interviene en la migración, diferenciación celular y organogénesis del embrión. Este gen JAG 1 codifica un ligando Notch 1 cuya expresión se relaciona con la embriogénesis de los órganos afectados en este síndrome, como el aparato cardiovascular, el hígado y los vasos sanguíneos (3,4,11). Este defecto cromosómico se hereda con carácter autosómico dominante, con penetrancia completa pero con expresión variable (3-5,11).
Se encuentran mutaciones en el gen JAG 1 en el 88% de los casos, mientras que un 7% presenta deleciones cromosómicas. Entre la mutaciones encontradas, 50% a 70% son esporádicas o de novo, mientras que 30% a 50% son hereditarias (14-16).
Objetivo
Describir el caso clínico de un niño con síndrome de Alagille, con afectación familiar, en el cual el diagnóstico se realizó en la etapa escolar, durante una hospitalización por patología aguda intercurrente.
Historia clínica
RM, 10 años, sexo masculino, raza blanca, procedente de Montevideo. Producto de primera gestación sin antecedentes perinatales patológicos. En los primeros años se diagnosticó estenosis pulmonar leve, no recibiendo tratamiento ni controles. Presentó una falla de crecimiento, con talla baja que no fue estudiada. Fracaso escolar, con dificultad en el aprendizaje. Se diagnosticó trastorno de conducta y se inició tratamiento con psiquiatra infantil.
Entre los antecedentes familiares se destaca: hermano de 5 años portador de cardiopatía congénita compleja, primas portadoras de cardiopatía congénita y dificultades en el aprendizaje.
El niño ingresó por una probable artritis séptica de cadera. Comenzó 4 días previos al ingreso con fiebre y agregó en la evolución coxalgia e impotencia funcional. Centellograma óseo compatible con artritis de cadera. Se realizó una artrocentesis, extrayéndose líquido con abundantes polimorfonucleares, sin desarrollo bacteriano. Se realizó drenaje quirúrgico y tratamiento antibiótico.
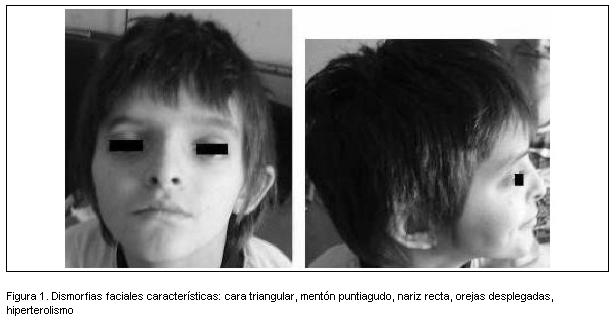
Se constataron dismorfias faciales: cráneo con predominio del diámetro antero-posterior, con aplanamiento occipital, cara triangular, hipertelorismo, ojos profundos, pabellones auriculares desplegados, narinas en anteversión. No se evidenciaron alteraciones morfológicas a nivel de miembros ni tronco. El examen pleuro-pulmonar era normal. Punta cardíaca en sitio de referencia, ritmo regular de 90 latidos por minuto, soplo sistólico, eyectivo, de intensidad 4 en 6 más intenso en foco pulmonar. Pulsos y presión arterial normales. Examen abdominal normal. No dolor a la palpación ni movilización de articulación de la cadera. Herida de sutura sin signos fluxivos.
Se continuó el tratamiento antibiótico por 25 días. La evolución fue buena, en apirexia, sin alteraciones en el examen ósteo-articular y sin otras complicaciones.
Desde el ingreso llamó la atención el fascies del paciente (figura 1), que además era muy similar al de su madre. Teniendo en cuenta los antecedentes de cardiopatía congénita, talla baja, dificultades en el aprendizaje y antecedentes familiares de cardiopatía congénita, se sospechó un probable síndrome genético, planteándose un síndrome de Alagille.
En vistas a confirmar el diagnóstico se realizó ecocardiograma, que confirmó la presencia de estenosis pulmonar sin repercusión hemodinámica.
La radiografía de columna mostró cuerpos y discos vertebrales sin alteraciones, con cierre incompleto de la primera vértebra sacra.
La valoración por médico oftalmólogo evidenció condensación a nivel de la córnea periférica correspondiente a embriotoxon.
Se realizó hemograma y funcional y enzimograma hepático sin alteraciones, al igual que perfil lipídico (tabla 1).

Ecografía abdominal sin hepatomegalia, con dos litiasis móviles de 5 y 10 mm a nivel de vesícula biliar.
En suma, en este paciente se diagnosticó síndrome de Alagille con cuatro criterios mayores: cardiopatía congénita, embriotoxon posterior, alteraciones vertebrales y dismorfias faciales. Presenta, además, dos anomalías asociadas: retardo mental y talla baja.
Debido a que no presenta alteraciones clínicas ni bioquímicas que orienten a patología hepática, por el momento no se plantea la realización de biopsia, que será considerada si la evolución clínica sugiere compromiso a ese nivel. Debe ser tenido en cuenta el hecho de que se ha demostrado un incremento en los hallazgos histológicos de paucidad ductal a medida que aumenta la edad del paciente (hasta en un 60% de niños y un 95% en mayores) (7).
Destacamos que nuestro paciente no asocia desnutrición severa (presente hasta en el 50% de los casos) (10). Esto podría deberse a la ausencia de compromiso hepático con malabsorción de ácidos grasos, así como a la ausencia de reflujo gastroesofágico.
La historia familiar de cardiopatía congénita en 2 primos y un hermano, además del fascies característico en varios familiares, sugieren la afectación familiar por esta enfermedad. Se realizó el familiograma que muestra la distribución característica de los trastornos con herencia autosómica dominante (figura 2). Cabe destacar que ninguno de los familiares tiene antecedentes sugestivos de patología hepática.
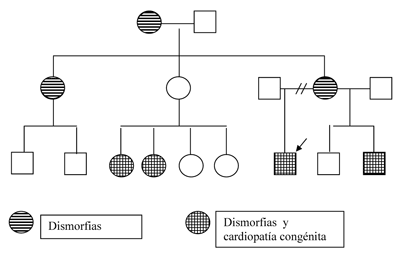
Figura 2. Árbol genealógico con características de trastorno autosómico dominante
Se coordinó el seguimiento del paciente y sus familiares por la Policlínica de Referencia del Centro Hospitalario Pereira Rossell, así como también fueron vinculados a las policlínicas de genética, cardiología infantil y gastroenterología.
Discusión
El síndrome de Alagille se caracteriza por dismorfias faciales típicas, acompañadas de anomalías hepáticas, esqueléticas, renales, oculares y cardiovasculares (1-7).
Los rasgos faciales característicos incluyen cara triangular, frente amplia y prominente, hipertelorismo, ojos profundos, nariz recta, puente nasal amplio con nariz recta y bulbosa, aplanamiento malar, maxilar inferior pequeño y mentón puntiagudo (2,7).
Las manifestaciones a nivel hepático son muy variables, desde una leve colestasis hasta una falla hepática progresiva. Estas variaciones se han visto incluso en la misma familia. La mayoría de los pacientes que tienen síntomas hepáticos tienen ictericia. Se presentan frecuentemente en los primeros 3 meses de vida con ictericia colestática, evidenciada por aumento de bilirrubina conjugada, ácidos biliares, fosfatasa alcalina, gamma glutamil transpeptidasa y triglicéridos. El síndrome de Alagille es una de las causas más frecuentes de colestasis neonatal (6-9).
La evolución de la enfermedad hepática también es muy variable. Aproximadamente el 15% de los afectados requieren trasplante hepático, representando el 2% de los trasplantes en la edad pediátrica (7,16).
El trastorno hepático se debe a la escasez de conductos biliares intrahepáticos. El diagnóstico de esta alteración se confirma mediante la biopsia hepática que evidencia la escasez de ductos, con una relación conductos biliares/espacios porta menor a 0,4. Esta alteración histopatológica es progresiva, y no es habitual verla en la etapa de recién nacido (6-9).
La mayoría de los portadores de síndrome de Alagille, hasta el 90%, presentan escasez de conductillos biliares (7,11). A pesar de que ésta se considera la manifestación más importante del síndrome, puede no estar presente en todos los afectados (3,7,11,17,18).
Dentro de las manifestaciones cardíacas, el soplo cardíaco está presente en un 85% de los casos. Los vasos pulmonares son los más frecuentemente afectados, siendo la cardiopatía más frecuente la estenosis valvular o subvalvular pulmonar, que aparece en el 67% de los casos. El defecto cardíaco complejo más frecuente es la tetralogía de Fallot que puede aparecer en 7% a 16% de los afectados. Otros defectos cardíacos menos frecuentes son defectos septales ventriculares, estenosis aórtica y coartación aórtica (7).
A nivel oftalmológico la manifestación más frecuente es el embriotoxon posterior que se encuentra entre el 78% y el 89% de los casos (7). Es una alteración de la cámara anterior del ojo, que se detecta al examen con lámpara de hendidura y que no afecta la agudeza visual. Esta alteración se puede observar en un 8% a 15% de la población sana. Otros hallazgos menos frecuentes son retinitis pigmentosa y afectación del iris por destrucción de la membrana de Descemet (7).
Entre 33% y 87% de los enfermos presenta malformaciones esqueléticas, como hemivértebras (vértebras “en mariposa”), hipoplasia de los cuerpos vertebrales, o alteraciones costales (2,7).
En aproximadamente un 50% de los casos se presenta desnutrición severa, considerada un signo clínico mayor para el diagnóstico. Ésta se vincula a la malabsorción de ácidos grasos debida a la colestasis así como a reflujo gastroesofágico (10).
Entre las anomalías menores que se han asociado a este síndrome, los xantomas son debidos a los altos niveles de colesterol. Algunos pacientes pueden presentar insuficiencia pancreática. Se ha reportado retardo mental en 30% de los casos. Se encuentra falla de crecimiento en 50% a 90% de los afectados (7). En algunos pacientes se han reportado accidentes cerebro vasculares (18) y craneosinostosis (3,7). Un 74% de los casos asocian patología renal, siendo los hallazgos más comunes proteinuria y quistes renales (7).
Este síndrome no tiene signos o síntomas patognomónicos (1-7,11), el diagnóstico se realiza por la asociación de manifestaciones clínicas. Los criterios diagnósticos clásicos, descriptos por Alagille en 1987 incluyen paucidad de ductos biliares en la biopsia hepática; junto a tres de los cinco criterios clínicos (2) (tabla 2).

Actualmente se plantea el diagnóstico con la presencia de cuatro o más criterios mayores en aquellos casos en que no se demostró paucidad ductal en la biopsia hepática, o en los que ésta no se realizó (20). Se denominan formas incompletas a las que presentan tres criterios y formas completas a aquellas con cuatro criterios mayores.
El diagnóstico puede confirmarse con el análisis de ADN en búsqueda de las alteraciones del gen JAG 1. Se detectan mutaciones hasta en un 70% de los casos (14).
El pronóstico de este síndrome depende de la entidad de la afectación hepática y las malformaciones cardiovasculares. Entre 21% y 31% de los pacientes requiere trasplante hepático, las principales indicaciones son insuficiencia hepatocítica, hipertensión portal o prurito intenso. Hasta un 15% puede desarrollar complicaciones como hepatocarcinoma, insuficiencia hepatocítica, insuficiencia pancreática exócrina, fibrosis hepática con hipertensión portal (7,8,12).
Referencias bibliográficas
1. Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr 1975; 86: 63–71.
2. Alagille D, Estrada A, Hadchouel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr 1987; 110: 195–200.
3. Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner N. Consequences of JAG1 mutations J Med Genet 2003; 40: 891–5.
4. Elmslie F V, Vivian A J, Gardiner H, Hall C, Mowat A, Winter R. Alagille Syndrome: family Studies. J Med Genet 1995; 32: 264-8.
5. Mueller RF. The Alagille Syndrome (Arteriohepatic dysplasia). J Med Genet 1987; 24: 621-6.
6. Watson GH, Miller V. Arteriohepatic dysplasia: familial pulmonary arterial stenosis with neonatal liver disease. Arch Dis Child 1973; 48: 459-66.
7. Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999; 29: 822–9.
8. Ruiz M, Michel F, Cervantes R, Zárate F, Mata N, Montijo E, et al. Síndrome de Alagille: informe de 12 casos en el Instituto Nacional de Pediatría. Rev Enf Infec Pediatr 2007; 81: 13-7.
9. Frauca E, Muñoz G. Colestasis en el lactante, En: Sociedad Española de Gastroenterolgía, Hepatología y Nutrición Pediátrica. Tratamiento en Gastroenterología, Hepatología y Nutrición Pediátrica. Madrid: Ergón, 2004; 433-447.
10. Roberts EA. The jaundiced baby, En: Kelly DA. Diseases of the liver and biliary system in children. 3 ed. Oxford: Blacwell Publishing, 2008: 73-6.
11. Spinner N, Krantz I, Kamath B. Alagille Syndrome [online]. Gene reviews 2000 may 19. Obtenido de: www. genetests.org [consulta: 3 mar. 2009]
12. Méndez MV, Scaglia J, Miralles M, Gutiérrez C, Ferrari AM. Síndrome de Alagille. Un caso de evolución poco habitual. Arch Pediatr Urug 1994, 65(3): 37–42.
13. Méndez MV, Burastero M, Cuadro MN, Casamayou L, Gutiérrez C, Montano A, et al. Colestasis neonatal: evolución de los pacientes diagnosticados entre 1983 y 2000 en dos servicios gastroenterológicos de referencia de Montevideo, Arch Pediatr Urug 2005; 76(3): 210-21.
14. Krantz I, Colliton P, Genin A, Rand E, Li L, Piccoli D, et al. Spectrum and Frequency of Jagged1 (JAG1) Mutations in Alagille Syndrome Patients and Their Families. Am J Hum Genet 1998; 62: 1361–9.
15. Colliton RP, Bason L, Lu FM, Piccoli DA, Krantz ID, Spinner NB. Mutation analysis of Jagged1 (JAG1) in Alagille syndrome patients. Hum Mutat 2001; 17(2): 151-2.
16. Boyer J, Crosnier C, Driancourt C, Raynaud C, Gonzales M, Hadchouel M et al. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Hum Genet 2005; 116: 445–53.
17. Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut 2001; 49: 431-5.
18. Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet 1997; 34(2): 152-7.
19. Kamath B, Spinner N, Emerick K, Chundley A, Booth C, Piccoli D, et al. Vascular Anomalies in Alagille Syndrome: A Significant Cause of Morbidity and Mortality. Circulation 2004; 109: 1354-8.
20. Wang JS, Wang XH, Zhu QR, Wang ZL, Hu XQ, Zheng S. Clinical and pathological characteristics of Alagille syndrome in Chinese children. World J Pediatr 2008; 4(4): 283-8.
Correspondencia: Dra. Elsa Arocena.
Correo electrónico: exbaloo@gmail.com.


{kind=link}