










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.80 no.2 Montevideo jun. 2009
CASO CLÍNICO
Arch Pediatr Urug 2009; 80(2)
Un fenotipo clínicamente reconocible: síndrome alfa-talasemia con retraso mental ligado al cromosoma X (ATR-X).
Análisis clínico y molecular en dos hermanos
Dres. Aída Lemes 1, Andrea Quadrelli 1, Cristina Ferolla 2,
Rosario Bonaglia 2, Mariela Larrandaburu 1, Alicia Vaglio 1, Roberto Quadrelli 1
1. Instituto de Genética Médica, Hospital Italiano, Montevideo.
2. Departamento de Neuropediatría, Banco de Previsión Social, Montevideo.
Correspondencia: Aída Lemes. Instituto de Genética Médica. Hospital Italiano. Bulevar Artigas 1632, ZP 11600. Montevideo, Uruguay. Correo electrónico: lemesa@adinet.com.uy
Fecha recibido: 18 de abril de 2009.
Fecha aprobado: 30 de julio de 2009.
Resumen
El síndrome alfa-talasemia con retraso mental (ATR-X) es un desorden recesivo ligado al cromosoma X que asocia en varones severo retardo del desarrollo y mental, dismorfias faciales características, malformaciones genitales y anemia. Generalmente, las portadoras femeninas son física e intelectualmente normales. Hasta el momento se han descrito alrededor de 168 pacientes. En el presente trabajo, se describen dos hermanos con las características clínicas típicas del síndrome ATR-X con algunas variaciones neurológicas y hematológicas entre ambos. El diagnóstico, originalmente propuesto por la similitud de las características dismórficas con los casos previamente publicados y los hechos genealógicos, se afirmó al demostrar inclusiones de hemoglobina HbH en glóbulos rojos luego de su incubación con azul de cresil brillante (BCB), y se confirmó mediante el análisis molecular del gen ATRX que mostró la mutación frecuente 736C>T. El presente trabajo tiene como objetivo alertar al pediatra sobre el ATR-X por tratarse de retardo mental sindromático en varones con un fenotipo clínico fácilmente reconocible. Hasta donde sabemos, estos son los primeros pacientes reportados con el síndrome ATR-X con definición molecular en América del Sur.
Palabras clave:
ALFATALASEMIA
RETRASO MENTAL LIGADO AL CROMOSOMA X
HEMOGLOBINA H
Summary
ATR-X syndrome is an X-linked disorder which affects boys. The syndrome is characterized by severe development delay and mental retardation, characteristic facial features, genital abnormalities, and anemia. Generally, female carriers are physical and intellectually normal. At present, about 168 patients have been reported. Here, we describe two brothers who have the ATR-X syndrome with typical clinical characteristics with some neurological and hematological variations between both of them. The diagnosis, originally suspected because of similarity in dysmorphic features to previous cases and the genealogical information, was defined haematologically with the demonstration of haemoglobin HbH inclusions in red blood cells after incubation with brilliant cresyl blue (BCB). Molecular analyses in the ATRX gene showed the common mutation 736C>T in both patients. The objective of the present work is to alert pediatrician to ATR-X syndrome which involves mental retardation in affected boys with a clinical phenotype easily recognized. To our knowledge, these are the first patients with molecular definition reported in South America.
Key words:
ALPHA-THALASSEMIA
MENTAL RETARDATION, X-LINKED
HEMOGLOBIN H
Introducción
El síndrome de ATR-X (alpha-thalassemia/mental retardation, X-linked) (OMIM 301040) es un desorden recesivo ligado al cromosoma X que afecta a pacientes de sexo masculino. Fue descrito por primera vez por Wilkie y colaboradores en el año 1991; si bien la rara asociación entre alfa-talasemia y retardo mental fue reconocida por primera vez por Weatherall y colaboradores en 1981, quienes propusieron un factor genético común responsable de las diversas manifestaciones clínicas (1,2). Posteriormente, se observó que la asociación entre alfa-talasemia y retardo mental está presente en dos síndromes diferentes: síndrome de genes contiguos por deleción en el brazo corto del cromosoma 16, donde se encuentra el gen de la alfa-globina (3), y en el síndrome ATR-X al cual nos referimos en este artículo.
Las características clínicas de los pacientes varones afectados por el síndrome ATR-X incluyen retardo mental severo, rasgos faciales clínicamente reconocibles durante la infancia, signos hematológicos de alfa-talasemia, malformaciones esqueléticas y genitales (4-6). Otros elementos fenotípicos incluyen microcefalia postnatal, baja talla, convulsiones, defectos cardíacos y malformaciones de la vía urinaria (5,7). Se desconoce la prevalencia en la población general; un estimativo para la prevalencia es <1-9/1.000.000 (5). Si bien, originalmente, la presencia de alfa-talasemia fue uno de los elementos centrales en la definición del síndrome dado que está presente en la mayoría de los niños (90%), existe una variación considerable en las manifestaciones hematológicas asociadas con las mutaciones en el gen ATRX (5). Una característica de la alfa-talasemia es la presencia de inclusiones de hemoglobina H en glóbulos rojos que les da una apariencia característica referida como en “pelota de golf” al microscopio óptico (8,9). Justamente, el test diagnóstico más sensible y muy sencillo para afirmar el diagnóstico de ATR-X es la demostración de inclusiones de hemoglobina H (exceso de cadenas beta) en glóbulos rojos luego de su incubación con azul brillante de cresilo (BCB) al 1% (10).
El síndrome es resultado de mutaciones en el gen ATRX, que codifica la proteína ATRX de amplia expresión (5). Esta proteína intervendría en la regulación de la expresión génica, entre otros, del gen de la alfa-globina. Se han encontrado mutaciones en el gen ATRX (localizado en Xq13.3) en aproximadamente la mitad de los pacientes afectados, reportándose hasta el momento más de 70 mutaciones distintas (8,10-12).
En el presente trabajo se describen dos hermanos con el típico fenotipo clínico pero con algunas diferencias en la presentación neurológica y hematológica del cuadro clínico. Los pacientes presentan facies típicas, retardo madurativo y mental, y anemia. Ambos tienen la mutación 736C>T en el gen ATRX. El objetivo de la presente publicación es alertar al pediatra de un fenotipo clínico fácil de reconocer en varones con retardo mental, el cual tiene un test sencillo para su definición.
Materiales y métodos
Paciente 1
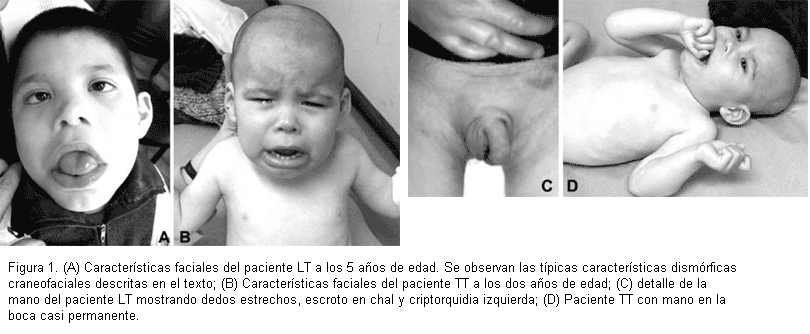
El propósito LT, con 6 años de edad al momento del diagnóstico, es producto del primer embarazo de pareja no consanguínea, de 17 años de edad la madre y 20 años el padre al momento del nacimiento. Parto de término. Los datos antropométricos al nacer fueron normales (tabla 1). A los 14 días de vida ingresó en sala de pediatría por mal progreso ponderal y dificultades de succión. A las 24 horas del ingreso presentó apnea por lo cual pasó a unidad de terapia intensiva egresando luego de 19 días de internación con diagnóstico de sepsis estafilocócica, convulsiones y soplo cardíaco en estudio. Durante el primer año de vida tuvo múltiples internaciones. Operado al año de vida de cardiopatía congénita (CIA) con buena evolución posterior. La madre refiere varias internaciones por anemia crónica (que requirió transfusión de glóbulos rojos en algunas oportunidades), desnutrición y raquitismo. Presenta retraso en la adquisición de pautas madurativas y retardo mental severo. Se encuentra en silla de ruedas. Tendría un vocabulario de cinco palabras. No tiene control esfinteriano. El paciente lleva las manos a la boca en forma casi constante. Desde los 3 años se encuentra en rehabilitación con foniatra y psicomotricista notando los padres lenta mejoría, especialmente en la deglución. Tiene escaso contacto ocular. Presenta problemas serios de constipación. Al examen físico presenta peso de 16 kg (percentil < 3), talla de 110 cm (percentil < 3) y perímetro cefálico de 45,5 cm (percentil < 3); cara redondeada, frente estrecha, remolino central en la línea de implantación anterior del cabello, occipucio plano, rebordes orbitarios poco marcados, hendiduras palpebrales pequeñas inclinadas hacia arriba, hipertelorismo ocular, epicanto bilateral, nistagmus horizontal de ojo izquierdo, punta nasal triangular con narinas antevertidas, micrognatia, boca grande con labios gruesos, labio inferior evertido, paladar alto, filtrum liso y largo, lengua grande y protruyente con babeo permanente; orejas incompletamente rotadas. En las manos, se observa fijación en flexión de primer articulación interfalángica de dedos anular y meñique bilateral, dedos que se afinan distalmente. Se observa criptorquidia izquierda y escroto en chal (figura 1A y C). El examen neurológico mostró hipotonia generalizada, no se obtienen reflejos osteotendinosos. Electroencefalograma y biopsia muscular normales. Por el fenotipo clínico se sospecha el síndrome de ATR-X que se afirma con la observación de inclusiones de hemoglobina H en glóbulos rojos luego de su incubación con BCB al 1%.
Paciente 2
TT es hermano varón del propósito, de dos años de edad al momento de la primera consulta. Es producto del tercer embarazo de la pareja (véase la genealogía en la figura 2). Parto normal a término. Datos antropométricos al nacer normales (tabla 1). La madre refiere diagnóstico de anemia que no requirió transfusión, y constipación crónica. Retraso en la adquisición de pautas madurativas. Al examen físico se observa dismorfias faciales moderadas, similares a las observadas en su hermano tales como epicanto, puente nasal bajo, macroglosia, boca grande con labios gruesos y babeo permanente (figuras 1B y D). Niño muy irritable que mantiene su mano en la boca de forma casi permanente. Perímetro cefálico 42 cm (< percentil 3). Tono muscular disminuido con reflejos osteotendinosos vivos simétricos. Hipotiroidismo congénito. La tomografía computada mostró discreta dilatación ventricular, cisternal y subaracnoidea con asociación de hipodensidad de la sustancia blanca bihemisférica a predominio periventricular. Se observaron inclusiones de hemoglobina H en glóbulos rojos luego de su incubación con BCB al 1%.
En la tabla 1 se resumen las observaciones clínicas relevantes de los dos pacientes en relación con el síndrome ATR-X.
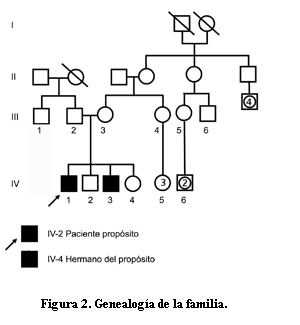
Análisis genealógico
El relevamiento genealógico muestra que los pacientes 1 (IV 1) y 2 (IV 3) son producto de primer y tercer embarazo y parto de pareja no consanguínea (figura 2). El segundo y cuarto embarazo finalizaron en un varón (IV 2) y en una niña (IV 4) clínicamente normales. No se detectan situaciones similares en la familia.
Análisis molecular
El análisis molecular fue realizado en colaboración con el Dr. Gibbons y colaboradores en la Unidad de Hematología Molecular de la Universidad de Oxford (Reino Unido) según se describe en Gibbons et al(…), 1995 (7,8).
Resultados
El análisis molecular en el gen ATRX, realizado en ambos hermanos, mostró una mutación frecuente 736C>T.
Discusión
El síndrome ATR-X es un desorden recesivo ligado al cromosoma X poco frecuente y generalmente muy severo en los varones afectados. Las características constantes en los pacientes con ATR-X son retardo mental y dismorfias faciales (9,13). En el presente trabajo describimos dos hermanos con el síndrome ATR-X. Al igual que en los casos previamente reportados, estos pacientes son varones con un severo retraso en su desarrollo, con retardo mental, una muy característica apariencia facial e historia de anemia crónica (tabla 1). Ambos pacientes fueron estudiados aplicando un test simple y rápido de búsqueda de inclusiones de HbH en eritrocitos luego de su incubación con BCB al 1%, el cual fue positivo en ambos. En este sentido, es importante destacar que, cuando por el fenotipo clínico y la historia familiar existe una fuerte sospecha de presencia del síndrome ATR-X, se debería realizar este test con la búsqueda cuidadosa de inclusiones de HbH y repetirlo ante un resultado inicial negativo. Dicho estudio debe realizarse con sangre fresca, recién obtenida, puesto que las inclusiones de HbH son inestables, por lo cual, las células con inclusiones pueden ser más difíciles de observar en muestras de sangre con más tiempo (5).
Como se indicó antes, el síndrome es resultado de mutaciones en el gen ATRX localizado en el brazo largo (q13.3) del cromosoma X (8,11,12). El gen ATRX codifica para una proteína nuclear similar a SWI/SNF (complejo remodelador de la cromatina). En los pacientes con el síndrome ATR-X se observan cambios en los patrones de metilación de las secuencias altamente repetidas. Se ha propuesto que la pérdida de función de la proteína ATRX puede estar relacionada con cambios en la estructura de la cromatina y de la metilación del ADN que conducen a la desregulación de la expresión génica, debido a que dicha proteína podría ser miembro de un gran complejo represor que incluya a las desacetilasas de histonas (5). Se postula que este mecanismo podría explicar la presencia de alfa-talasemia en estos pacientes que, como se indicó antes, no presentan mutaciones en el gen de la alfa-globina. Aproximadamente en la mitad de los pacientes con síndrome ATR-X se encuentran mutaciones en el gen ATRX (8,11,12). En los dos pacientes que aquí se describen, el análisis molecular en dicho gen identificó la mutación frecuente 736C>T, definida como una mutación de cambio de sentido (missense). Se trata de la sustitución de un único nucleótido en una secuencia de ADN que causa la sustitución de un aminoácido por otro en el producto génico y, como consecuencia, una alteración en la función normal de la proteína. En el caso del síndrome ATR-X, las mutaciones de cambio de sentido están localizadas en una región específica; el 50% a 60% de las mutaciones identificadas se encuentran en el dominio ADD, el 20% tiene la mutación 736C>T (que se encuentra en los pacientes que aquí se describen) y la siguiente región del gen frecuentemente afectada es la que corresponde al dominio de la helicasa (5).
Desde el punto de vista clínico, enfatizamos la variabilidad en la expresión clínica del síndrome ATR-X en ambos hermanos; así, mientras el hermano mayor presenta hipotonía con ausencia de reflejos osteotendinosos, lo que determinó (según datos de la historia clínica) la realización de una biopsia muscular por planteo de una posible enfermedad mitocondrial, el hermano menor presenta hipotonía con reflejos vivos. La severidad de la anemia también sería diferente, siendo significativamente mayor en el propósito que recibió transfusión de glóbulos rojos en varias oportunidades. La variabilidad en la expresión clínica de una enfermedad en hermanos es un hecho interesante porque a pesar de que ambos presentan la misma mutación, es posible que el efecto de la proteína ATRX en la expresión del gen de la alfa-globina se encuentre sujeto a factores genéticos y no genéticos diferentes en ambos hermanos. También la variabilidad observada en el fenotipo neurológico podría explicarse por la acción de otros genes, diferentes en ambos hermanos. Asimismo, destacamos que tanto las dismorfias faciales como la microcefalia postnatal fueron los elementos clínicos relevantes para proponer el diagnóstico de síndrome ATR-X en el propósito. En este sentido, resaltamos la importancia que tiene para el clínico el fenotipo característico de este síndrome, especialmente sus rasgos faciales distintivos en relación al gran capítulo de retardo mental ligado al cromosoma X. Por otra parte, y como ya indicamos, el test para alfa-talasemia es un test simple y de bajo costo que, cuando resulta positivo, permite afirmar el diagnóstico (5). En la esfera gastroenterológica es frecuente que los pacientes presenten vómitos recurrentes y/o reflujo gastroesofágico, especialmente en la niñez temprana (14); los pacientes reportados aquí no presentan historia de vómitos. La constipación se relata como hallazgo frecuente, al igual que en nuestros pacientes, demostrándose en algunos casos hipoganglionosis (14). Esto último no ha sido aún evaluado en ninguno de los hermanos.
A modo de conclusión, en este trabajo describimos dos hermanos con fenotipo clínico típico del síndrome ATR-X con inclusiones de HbH en eritrocitos y una mutación frecuente en el gen ATRX. Se destaca la importancia del diagnóstico molecular para confirmar el diagnóstico, así como para la identificación de las mujeres portadoras y la realización de asesoramiento genético familiar.
En la familia presentada, se justificaría realizar despistaje de portadora en la hermana de los afectados (IV 4), así como en la tía vía materna (III 4) y la prima de la madre (III 5). A las portadoras, es posible ofrecerle diagnóstico prenatal y/o preimplantacional de esta enfermedad.
De acuerdo a la bibliografía consultada, los pacientes que presentamos serían los primeros casos reportados con el síndrome ATR-X con confirmación molecular en América del Sur.
Agradecimientos
A Richard Gibbons y Chris Fisher de la Unidad de Hematología Molecular del Instituto Weatherall de Medicina Molecular (Hospital John Radcliffe, Universidad de Oxford) por la realización de los estudios moleculares. A la Dra. Verónica Nieto por la realización de estudios hematológicos. Agradecemos a los pacientes y sus familiares por su amable participación y contribución a este trabajo.
Referencias bibliográficas
1. Wilkie AO, Pembrey ME, Gibbons RJ, Higgs DR, Porteous ME, Burn J, et al. The non-deletion type of alpha thalassaemia/mental retardation: a recognisable dysmorphic syndrome with X linked inheritance. J Med Genet 1991; 10: 724.
2. Weatherall DJ, Higgs DR, Bunch C, Old JM, Hunt DM, Pressley L, et al. Hemoglobin H disease and mental retardation: a new syndrome or a remarkable coincidence? N Engl J Med 1981; 305(11): 607-12.
3. Wilkie AO, Zeitlin HC, Lindenbaum RH, Bucle VJ, Fischel-Ghodsian N, Chui DH, et al. Clinical features and molecular analysis of the alpha thalassemia/mental retardation syndromes. II. Cases without detectable abnormality of the alpha globin complex. Am J Hum Genet 1990; 46: 1127-40.
4. Gibbons RJ, Wilkie AO, Weatherall DJ, Higgs DR. A newly defined X linked mental retardation syndrome associated with alpha thalassaemia. J Med Genet 1991; 28(11): 729-33.
5. Gibbons RJ. Alpha thalassaemia-mental retardation, X linked. Orphanet J Rare Dis 2006; 1(15): 1-9.
6. Wada T, Sakakibara M, Fukushima Y, Sayito S. A novel splicing mutations of the ATRX gene in ATR-X syndrome. Brain Dev 2006; 28(5): 322-5.
7. Wilkie AO, Gibbons RJ, Higgs DR, Pembrey ME. X linked alpha thalassaemia/mental retardation: spectrum of clinical features in three related males. J Med Genet 1991; 28(11):738-41.
8. Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 1995; 80(6): 837-45.
9. Gibbons RJ, Brueton L, Buckle VJ, Burn J, Clayton-Smith J, Davison BC, et al. Clinical and hematologic aspects of the X-linked alpha-thalassemia/mental retardation syndrome (ATR-X). Am J Med Genet 1995; 55(3): 288-99.
10. Jones KL. Smith's recognizable patterns of human malformation. Philadelphia: Elsevier Saunders, 2006.
11. Cardoso C, Lutz Y, Mignon C, Compe E, Depetris D, Mattei MG, et al. ATR-X mutations cause impaired nuclear location and altered DNA binding properties of the XNP/ATR-X protein. J Med Genet 2000; 37: 746-751.
12. Giuliano F, Badens C, Richelme C, Levy N. ATR-X syndrome: a new mutation in the XNP/ATRX gene near the helicase domain. Arch Pediatr 2005; 12(9): 1372-5.
13. Villard L, Bonino MC, Abidi F, Ragusa A, Belougne J, Lossi AM, et al Evaluation of a mutation screening strategy for sporadic cases of ATR-X syndrome. J Med Genet 1999; 36: 183-186.
14. Martucciello G, Lombardi L, Savasta S, Gibbons RJ. Gastrointestinal phenotype of ATR-X syndrome. Am J Med Genet 2006; 140(11): 1172-6.
Correspondencia: Dra. Aída Lemes
Instituto de Genética Médica. Hospital Italiano.
Bvar. Artigas 1632. Montevideo.
Correo electrónico: lemesa@adinet.com.uy


{kind=link}
{kind=link}