










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.79 no.4 Montevideo dic. 2008
ARTÍCULO ORIGINAL
Arch Pediatr Urug 2008; 79(4)
Moyamoya en la población infantil no asiática.
Ampliación de casuística
Gabriel González 1, Ma Eugenia Russi 1, Jaume Campistol 2, Gonzalo Costa 3,
Ramón Navarro Balbuena 4, Roberto Crosa 3, Antony Capdevila 5, Teresa Solá 6
1. Servicio de Neurología Centro Hospitalario Pereira Rossell.
2. Servicio de Neurología Hospital San Joan de Dèu.
3. Servicio de Neurocirugía Centro Hospitalario Pereira Rossell.
4. Servicio de Neurocirugía Hospital San Joan de Dèu.
5. Unidad Diagnóstico por la Imagen Hospital San Joan de Dèu.
6. Angiografía Intervencionista Hospital San Joan de Dèu.
Hospital San Joan de Dèu- Hospital General de Catalunya. Servicio de Neurología y Neurocirugía. Universidad de Barcelona. España.
Centro Hospitalario Pereira Rossell. Cátedra de Neuropediatría-Neurocirugía. Montevideo-Uruguay.
Fecha recibido: 29 de julio de 2008.
Fecha aprobado: 10 de diciembre de 2008.
Resumen
La enfermedad o síndrome de moyamoya es una rara afección que provoca una oclusión de las carótidas intracraneales y determina una red vascular colateral anormal en la base del cráneo y que, por su aspecto arteriográfico, hace ya más de 50 años un grupo de neurocirujanos japoneses denominaron moyamoya, y de la cual persisten muchas interrogantes sin resolver.
Se realizó un estudio descriptivo y longitudinal de 19 niños con síndrome o enfermedad de moyamoya. La edad promedio de inicio fue de seis años. Del total de niños incluidos, doce debutaron en forma aguda con infarto cerebral; dos pacientes (con antecedentes de epilepsia y retraso) lo hicieron, uno con infarto y otro con hemorragia cerebral, otros dos con epilepsia y retraso, uno con trastorno del lenguaje, siendo en los dos restantes un hallazgo casual. Entretanto doce pacientes se consideraron idiopáticos (portadores de enfermedad de moyamoya) y siete asociados a enfermedades sistémicas (síndrome de moyamoya). El diagnóstico inicial se confirmó por angiorresonancia en diez casos y por arteriografía convencional en nueve. El seguimiento promedio fue de cinco años, once evidenciaron progresión clínica y ocho estabilidad, con un seguimiento promedio de siete y dos años respectivamente. Todos mostraron progresión angiográfica. En siete niños se realizaron diez cirugías de revascularización sin complicaciones y con buenos resultados clínicos y angiográficos. Dos niños fallecieron (uno por hemorragia cerebral y otro por un infarto expansivo) y catorce presentaron secuelas leves a moderadas. Resulta importante identificar los niños que se pueden beneficiar con la cirugía de revascularización, una de las pocas opciones terapéuticas para evitar la progresión y complicaciones de esta grave enfermedad.
Palabras clave:
ENFERMEDAD DE MOYAMOYA
ANGIOGRAFÍA CEREBRAL
REVASCULARIZACIÓN CEREBRAL
ISQUEMIA ENCEFÁLICA
Summary
Moyamoya's disease or syndrome is a rare illness wich causes symptoms of cerebral ischemia due to intracranial arteries' stenosis, with secondary abnormal vasculature networks at the base of the brain. Since the first description of moyamoya disease done by a group of Japanese neurosurgeons more than 50 years ago, this rare illness is still considered an intriguing disease.
The clinical features, treatment, imaging findings and outcomes of a series of not Asian children with Moyamoya disease are described in this study. A total of 19 six year old patients were analyzed. The initial presentation in 12 patients was infarctions, 2 with epilepsy and mental retardation, another 2 who had personal history of epilepsy and mental retardation developed symptoms of ischemic stroke (one of them), and hemorrhagic stroke the other one; 1experienced language problems, while in the last two the diagnosis was casual. On the other hand, 12 patientes were considered idiopathic cases (moyamoya's disease) and 7 were related to systemic illnes (moyamoya's syndrome). Initial diagnosis was achieved by magnetic resonance angiography in 10 cases, and conventional angiography in 9. All patients were followed up for a mean period of 5 years. While 11 patients experienced clinical worsening of symptoms, 6 asymptomatic. However, all of them showed angiographic progression. Seven patients underwent bypass surgery with no further complications, 2 passed away and 14 remained with moderate handicaps. A number of surgical procedures have been developed for revascularization, improving the outcome of some of these patients by preventing ischemic and hemorrhagic stroke. Due to the fact that moyamoya disease is not a silent disorder and its progress cause complications, surgical revascularization should always be considered in the management of these patients.
Key words:
MOYAMOYA DISEASE
CEREBRAL ANGIOGRAPHY
CEREBRAL REVASCULARIZATION
BRAIN ISCHEMIA
Introducción
Más de 50 años han transcurrido desde que un grupo de neurocirujanos japoneses (Shimizu, Takeuchi, Suzuki y Takaku) comunicaran esta extraña afección que provocaba la oclusión espontánea y lentamente progresiva de las arterias del polígono de Willis (especialmente en el primer tramo intracraneal de las carótidas) dando una red vascular anormal de pequeños vasos en la base cerebral; y a la cual denominaran por su aspecto angiográfico moyamoya, que en japonés significa “niebla o humo” (1-3).
Dada la variedad de enfermedades reconocidas en la actualidad, congénitas o adquiridas, que pueden asociar un cuadro angiográfico similar, se las ha denominado a estas últimas síndrome de moyamoya (SMM), reservando el nombre enfermedad de moyamoya (EMM) para las formas idiopáticas (4,5).
La forma de presentación más común en la edad pediátrica es el accidente cerebrovascular (ACV) encontrado en aproximadamente el 80% de los casos, y fundamentalmente de naturaleza isquémica (a diferencia del adulto que suele debutar con hemorragia). No obstante otras presentaciones menos frecuentes han sido descritas como convulsiones, cefaleas y movimientos anormales (5).
Los ACV en los niños son infrecuentes y generalmente están asociados a múltiples factores de riesgo. En su serie, Gabrielle de Veber reporta que el 50% de los ACV isquémicos arteriales presentan una vasculopatía, siendo la angiopatía posvaricela, la disección de los vasos intra y extracraneales, el moyamoya y las vasculitis las más frecuentes (6). Este motivo junto al mayor acceso a técnicas de imagen menos invasivas como la angiorresonancia (ARM), han determinado que esta entidad se identifique con mayor frecuencia. En Japón es la causa más frecuente de ACV en la infancia, mientras que en Occidente algunos trabajos indican que representa de 6% al 8% de la patología cerebrovascular infantil (5,6). Si bien las comunicaciones e investigaciones en relación al tema se han incrementado en las últimas décadas, aún existen muchos interrogantes en lo que a su etiopatogenia, evolución natural y diferencias raciales respecta; datando las primeras comunicaciones en español de la década del 70 (7-10).
Si bien la opinión de expertos y la revisión bibliográfica sugieren que la cirugía de revascularización directa y/o indirecta es la mejor opción terapéutica, y que la mayoría de los niños con moyamoya se beneficiarían con el tratamiento, la indicación, oportunidad, técnica quirúrgica y el impacto sobre la evolución natural de la enfermedad no están aún totalmente aclarados (11).
Motivan la realización de este trabajo las controversias que existen en las diferencias entre la población nipona y occidental, la evolución natural y la conducta terapéutica a adoptar, siendo el principal objetivo del mismo la descripción, presentación y evolución clínico-radiológica de niños no asiáticos con síndrome o enfermedad de moyamoya, mediante la ampliación de una casuística del autor (12).
Pacientes y métodos
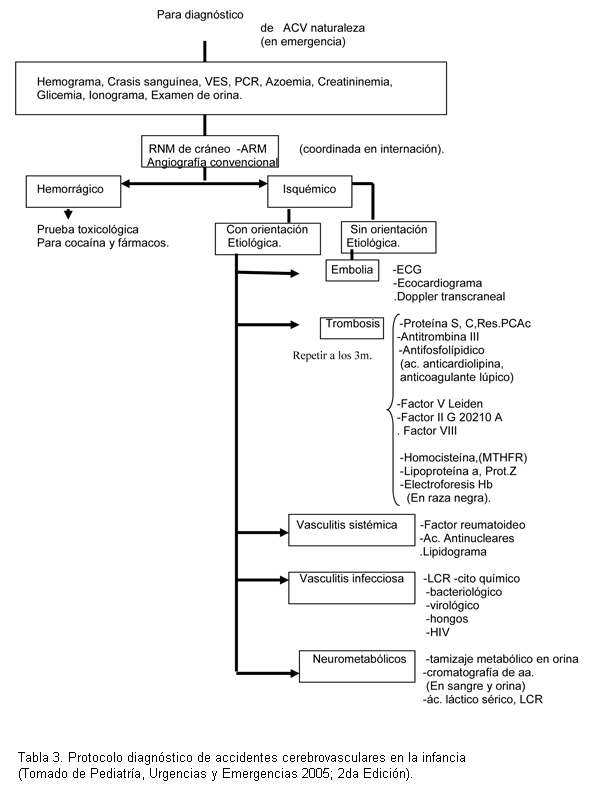
Se diseñó un trabajo descriptivo y longitudinal, analizando las historias clínicas de niños de 0 a 18 años atendidos en el Hospital Sant Joan de Dèu de Barcelona (HSJD) y el Centro Hospitalario Pereira Rossell (Policlínica de accidentes vasculares), en el período comprendido entre el 1 de junio de 1997 y el 1 de junio de 2007, con diagnóstico de síndrome o enfermedad de moyamoya de acuerdo a los criterios diagnósticos del Ministerio de Salud y Bienestar del gobierno japonés (13).
Se diseñó un protocolo de recolección de datos que incluía antecedentes familiares y personales, clínica, estudios complementarios, tratamiento y evolución.
Se incluyeron 19 niños en los cuales los estudios de ARM y/o angiografía evidenciaron cambios esteno-oclusivos en las porciones intracraneales terminales de las carótidas internas con desarrollo de vasos moyamoya en la base del cerebro. Si estas alteraciones eran bilaterales y no asociadas a otras enfermedades congénitas o adquiridas se consideraron como EMM. En cambio cuando las alteraciones eran asociadas a enfermedades congénitas o adquiridas se incluyeron como SMM.
Finalmente si eran unilaterales se denominaron síndrome o enfermedad de moyamoya unilateral.
La presentación clínica se clasificó en base al tiempo de evolución de los síntomas de la enfermedad al momento del diagnóstico en: aguda (menor de 15 días); subaguda (de 16 a 60 días) y crónica progresiva (más de 61 días). Los principales hallazgos clínicos son:
- ACV (AIT, infarto cerebral, hemorragia cerebral).
- Epilepsia.
- Retraso en el desarrollo.
- Trastorno del lenguaje.
- Movimientos anormales.
- Cefaleas.
- Hallazgo accidental.
Todos los pacientes contaron con estudios de neuroimagen que incluían resonancia magnética (RM) craneal con ARM y/o angiografía convencional.
Se clasificaron en base a los resultados en:
- Estadios de Houkin por ARM (14).
- Estadios de Suzuki por angiografía por punción (15).
Se analizó el tipo de tratamiento quirúrgico realizado y la evolución clínica en base a la escala de seguimiento de ACV de Rankin (16) y en pacientes operados según criterios de Kim (17) y de Matsushima (18).
Resultados
Se incluyeron diecinueve pacientes (n=19), de los cuales doce eran varones y siete niñas, con una relación estimada niños/niñas de 1,7/1. La edad de inicio fue de tres meses a quince años, con una media de seis años (figura 1).

La presentación clínica fue aguda en doce casos (fr: 0,63), crónica progresiva en cinco (fr: 0,26) y de hallazgo casual en dos (fr: 0,11) (tabla 1). En función del diagnóstico inicial se clasificaron en:
- Infarto cerebral (n=12).
- Infarto cerebral en niño con retraso y epilepsia (n=1).
- Hemorragia cerebral en un niño con retraso y epilepsia (n=1).
- Trastorno del lenguaje (afasia) (n=1).
- Epilepsia y retraso (n=2).
- Sin síntomas (n=2).
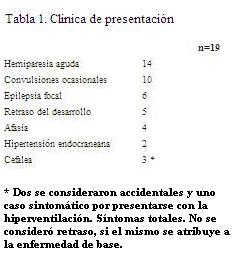
Cuatro niños (fr: 0,21) con infarto cerebral tuvieron antecedentes de accidente isquémico transitorio relacionado con la hiperventilación. Los dos casos asintomáticos encontrados presentaron historia típica de cefaleas primarias (migrañosa y tensional). Doce casos fueron idiopáticos (fr: 0,63) (EMM) y siete (fr: 0,37) sintomáticos (SMM) secundarios a: síndrome de Down (tres casos), neurofibromatosis tipo 1 (NF1), drepanocitosis, arteriopatía sistémica idiopática y síndrome de Simpson-Golabi-Behmel. Dieciséis casos fueron bilaterales (fr: 0,84) y tres (fr: 0,16) inicialmente unilaterales; de éstos dos desarrollaron en la evolución una alteración bilateral. De los 16 pacientes a los que se les realizó EEG con hiperventilación, tan sólo seis presentaron EEG con fenómeno “re build-up”, que consiste en ondas lentas de alto voltaje, irregulares y baja frecuencia con la hiperventilación.
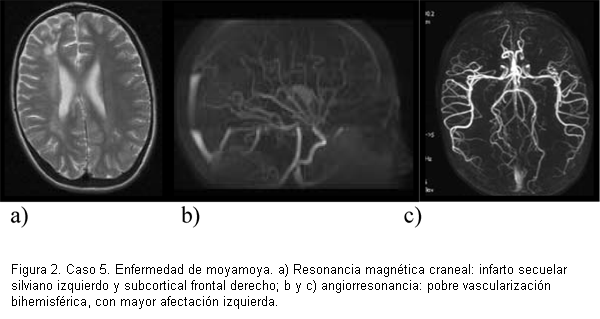
La TAC o la RM craneal inicial en diecisiete casos evidenció zonas de isquemia: nueve fueron isquemias agudas, cuatro agudas y secuelares y cuatro secuelares (figuras 2 y 3). En un caso se encontró hemorragia mientras que el otro fue asintomático unilateral.
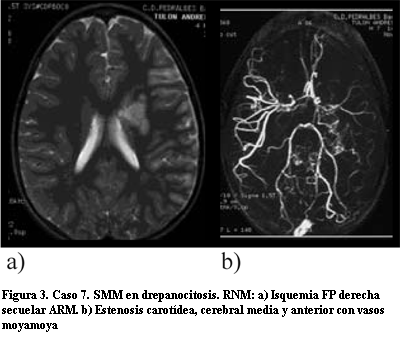
El diagnóstico inicial fue realizado por ARM en diez casos y por arteriografía convencional en nueve (tabla 2) (figuras 2, 3 y 4). En seis pacientes la ARM inicial no confirmó el diagnóstico informando: oclusión cerebral media en tres casos, hipoplasia carotídea en uno, probable angiomatosis leptomeníngea en dos y estudio normal en los otros dos restantes. En un solo caso se realizó SPECT que demostró zonas de hipoperfusión.
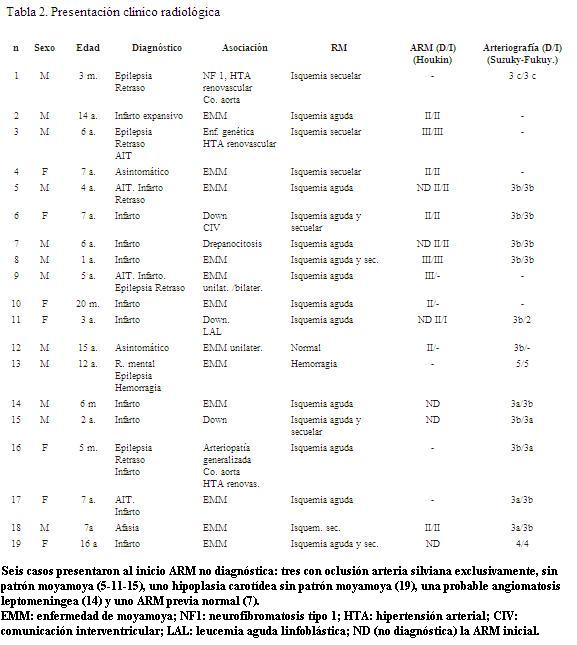
Todos los pacientes fueron estudiados según protocolo de accidentes cerebrovasculares (tabla 3), encontrando en un caso drepanocitosis (caso 7) y en otro trombocitosis que no reiteró en la evolución (caso 10).
El tiempo de seguimiento clínico fue de 19 años, con una media de 5 años.
La evolución a nivel neurológico evidenció progresividad en once casos (nueve repitieron accidentes cerebro vasculares isquémicos y/o hemorrágicos) y estabilidad en los ocho restantes. La media de seguimiento en los casos de progresividad clínica fue de siete años versus dos años en los casos de estabilidad (tabla 4).
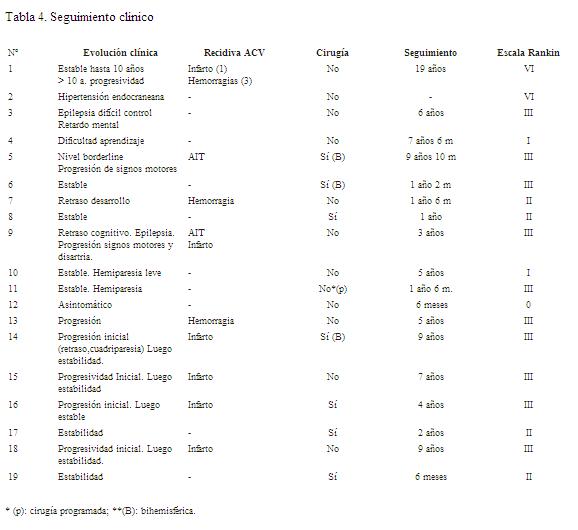
El seguimiento angiográfico no pudo establecerse en siete pacientes (dos fallecieron sin nuevos controles, dos fueron intervenidos en un intervalo menor de seis meses, con controles posteriores pendientes, y en tres no fueron solicitados). En los doce restantes se evidenció progresión de la enfermedad (tabla 5).
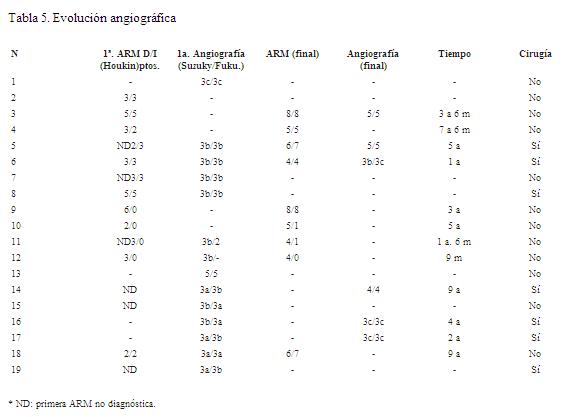
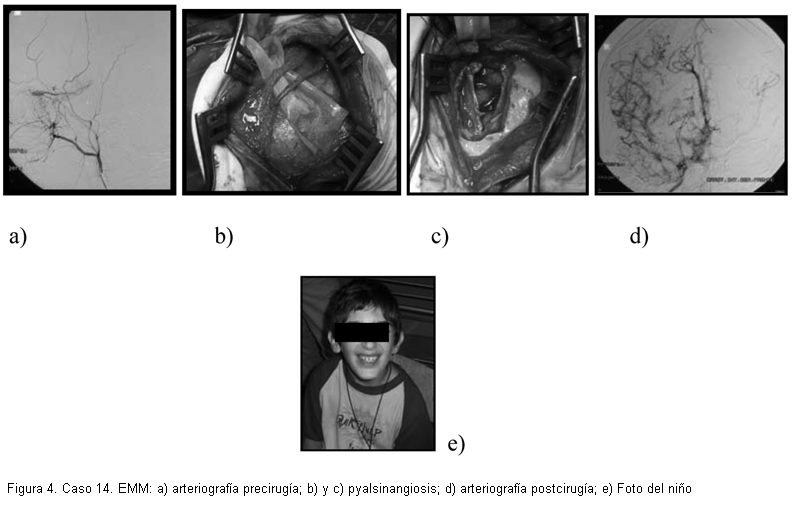
En cuanto al tratamiento, se realizaron diez cirugías de revascularización indirecta en siete niños (tres bihemisféricas), sin complicaciones perioperatorias (figura 4). En las diez cirugías se realizó encefaloduroarteriosinangiosis (EDAS) modificada: nueve pialsinangiosis y una modificada con trépano frontal (caso 8). De los diecinueve pacientes, quince recibieron ácido acetil salicílico (100 mg/día) y nueve calcioantagonistas (flunaricina 5-10 mg/día). Los cuatro casos con hipertensión renovascular recibieron antihipertensivos.
La evolución clínica en los operados (según escala de Kim) fue aceptable (grado 1) en un caso, buena (grado 2) en cinco casos y aún no establecida en otro caso, por evolución inferior a seis meses. En cuanto a la evolución radiológica en las diez cirugías efectuadas fue aceptable, según los criterios de Matsushima (grado 2) en un caso, buena (grado 1) en siete casos, estando en los dos restantes pendientes los controles angiográficos.
Dos pacientes fallecieron, el caso 1 por hemorragia cerebral y el 2 por infarto expansivo con hipertensión endocraneana incontrolable.
Discusión
La enfermedad de moyamoya se caracteriza por la oclusión espontánea y lentamente progresiva de las arterias del polígono de Willis, con la aparición simultánea de colaterales intracerebrales y extracraneales, de etiología poco clara.
Si consideramos las formas idiopáticas (EMM), la mayor frecuencia se da en la población asiática (especialmente en Japón y Corea), con una incidencia en estas regiones de 3,16 casos/100.000 habitantes/año, siendo 10 veces más frecuente que en Europa (4,5). Los casos reportados procedentes de países no asiáticos, al igual que en nuestra casuística, evidencian un alto número de casos sintomáticos vinculados a enfermedades sistémicas, siendo el síndrome de Down, la neurofibromatosis tipo I, la radiación craneal y las cardiopatías congénitas corregidas, las causas más frecuentes (5).
Esta diferencia podría explicarse por la no inclusión en general de casos de moyamoya sintomáticos en las comunicaciones orientales.
En nuestra serie dentro de los casos sintomáticos predominó el síndrome de Down, que presenta 30 veces más riesgo que la población general de desarrollar SMM (19). Cuatro asociaron más de un factor de riesgo como coartación de aorta, cardiopatía congénita y displasia arterial renal con hipertensión, elementos considerados de mal pronóstico.
Su etiopatogenia permanece hoy día sin aclarar, encontrándose a nivel anatomopatológico un engrosamiento fibrocelular de la íntima con adelgazamiento de la capa muscular media, sin cambios inflamatorios. Se sospecha la presencia de factores genéticos, con un 10% de casos familiares y otros factores ambientales no bien dilucidados hasta el momento actual (4,5,7). La NF1 es un ejemplo clásico donde se sabe existen factores genéticos (por mutación del gen que codifica la neurofibromina (17q11.2), que se expresarían a nivel del endotelio y músculo liso vascular y sobre el cual ciertos factores ambientales, como la radioterapia, incrementarían aún más el riesgo de SMM (20).
El incremento del factor básico de crecimiento fibroblástico (bFGF) en LCR observado en estos pacientes entretanto sería un marcador del aumento de la angiogénesis. En tal sentido, los últimos trabajos clínico- patológicos han encontrado una sobreexpresión del factor hipoxia-inducido (HIF-1alfa) en los vasos cerebrales de pacientes con EMM, lo cual podría afectar la síntesis de elastina (8,9).
La presentación clínica más frecuente en nuestra serie fue la hemiparesia aguda con o sin convulsiones ocasionales (como expresión de un infarto cerebral), observada en doce casos. Es de destacar que la presentación insidiosa o progresiva suele ser más frecuente en la infancia, como lo demuestra nuestra casuística, en la que cinco casos presentaron retraso del desarrollo, epilepsia y trastornos del lenguaje. En la serie de Smith y Scooth por otro lado, sobre 143 casos (adultos incluidos) predominó la isquemia en un 67% y las convulsiones en el 6,3% (5).
Cuatro casos presentaron al inicio accidentes isquémicos transitorios con la hiperventilación, lo cual sería secundario a hipocapnia que disminuye el flujo sanguíneo cerebral y determina isquemia, siendo frecuentemente catalogados en forma errónea como episodios de mecanismo epiléptico.
En nuestra casuística, diez niños presentaron convulsiones y seis desarrollaron epilepsia, uno de ellos de difícil control. Se estima que aproximadamente 50% de los pacientes presentan convulsiones en la fase aguda de la patología vascular infantil, con 48% que desarrollan epilepsia (siendo la mitad de ellas de difícil control) (21).
El caso 2 presenta un infarto con transformación hemorrágica de comportamiento expansivo que le provoca la muerte (siendo excepcional esta forma de presentación en la infancia), existiendo publicaciones que evidencian la eficacia de la hemicraniectomía decompresiva ante la hipertensión endocraneana refractaria al tratamiento médico, evitando la muerte con menores secuelas en niños (22,23). La presentación hemorrágica como la encontrada en el caso 13, no obstante, es excepcional en el niño, observando en general la típica curva de expresión bimodal con una presentación isquémica más frecuente en niños y hemorrágica en adultos (por el sangrado de vasos colaterales profundos). El caso 1 ejemplifica este comportamiento presentando ACV isquémico en la infancia y falleciendo en la edad adulta por una hemorragia cerebral.
En dos niños el diagnóstico fue casual al practicar una neuroimagen por cefaleas, siendo la presentación exclusiva con cefaleas excepcional y difícil de determinar si son sintomáticas o accidentales, reportándose en un reciente trabajo sobre 241 niños estudiados por cefaleas primarias tan sólo en dos casos (0,8%) de EMM (24).
En otra publicación del grupo coreano de Seol sobre 204 niños con EMM, encuentran un 21% con cefaleas preoperatorias en asociación en casi todos los casos con AIT e hiperventilación (25). Estos autores demuestran que la cefalea puede persistir o aparecer luego de la cirugía y plantean que la disminución o redistribución del flujo sanguíneo cerebral podría ser la causa de las mismas en estos pacientes (26).
En nuestra serie, en seis casos el EEG evidenció el signo característico de ondas lentas alto voltaje y baja frecuencia con la hiperventilación, que es debido a la disminución del flujo sanguíneo cerebral regional. Este signo según otros trabajos estaría presente en el 75% de los casos y desaparecería con la cirugía. El menor porcentaje en nuestra serie (37,5%) podría explicarse por la falta de colaboración del niño en ausencia de una búsqueda exhaustiva por el técnico (7).
Actualmente en niños la ARM es aceptada por el “Comité de Obstrucción Espontánea del Círculo de Willis del Ministerio de Salud Japonés” como herramienta de diagnóstico y seguimiento en la EMM (4,24). Entretanto la arteriografía convencional tiene una mejor definición de imagen siendo de enorme valor diagnóstico y terapéutico en vistas a una posible cirugía. Seis pacientes estudiados por el evento isquémico tuvieron una ARM previa que no evidenciaba obstrucción carotídea ni fenómeno de moyamoya, lo cual puede ser explicado por la posterior progresión de la enfermedad o por limitaciones de la técnica debidas a una menor sensibilidad (lo cual debe ser considerado al evaluar un niño con ACV con ARM no concluyente). La angiografía es imprescindible para valorar los circuitos anastomóticos (y por lo tanto necesaria para la cirugía) ya que la ARM por sí sola no puede valorarlos (por ser aquel un fenómeno dinámico), no pudiendo precisar los calibres de las diferentes ramas de la carótida externa.
Todos nuestros pacientes, a excepción del caso que debutó con hemorragia, se presentaron en etapas grado II y III de Houkin y 3 de Suzuki. A propósito de lo cual es importante destacar los inconvenientes de la clasificación de Suzuki por:
- Su alto grado de subjetividad.
- La mayoría de los niños al realizarse el diagnóstico están en fase 3.
- No tiene gran valor pronóstico.
- Los grados 1 y 6 no tendrían vasos moyamoya y de este modo no cumplirían los criterios diagnósticos.
Clásicamente se considera un mayor riesgo de isquemia en la etapa II-III y mayor riesgo de hemorragia en la etapa IV-V, lo cual corroboramos en nuestra serie (7).
Actualmente Houkin propone una clasificación muy útil fundamentalmente para niños, en base a los hallazgos de ARM (14).
Tres de nuestros pacientes inicialmente se presentaron como moyamoya unilateral, dos en la evolución desarrollaron una forma bilateral (lo cual está descrito en esta afección). En la serie de Smith, entretanto, de dieciséis pacientes con forma unilateral, nueve desarrollaron bilateralidad en un período de 6 meses a 4 años (5).
En uno solo de nuestros pacientes se evaluó la perfusión cerebral por SPECT.
La tomografía por emisión de positrones (PET) y la tomografía por emisión de fotón único (SPECT), tienen un enorme valor para el estudio del flujo sanguíneo cerebral, así como para establecer la topografía de irrigación reducida y el riesgo de infarto (aspectos estos fundamentales al momento de decidir una conducta quirúrgica). El SPECT basal y con acetazolamida, que al provocar vasodilatación puede evidenciar zonas hipoperfundidas no evidentes en la primer imagen basal, se ha utilizado para establecer el diagnóstico y pronóstico al decidir indicación y oportunidad de una cirugía, así como parámetro de control de la eficacia de la revascularización (26).
El pronóstico de la enfermedad es difícil de establecer, porque la evolución natural es variable y no totalmente aclarada. La arteriopatía es progresiva, sin embargo en función de la edad, del grado de severidad y del desarrollo de colaterales, la evolución puede ser muy lenta, con largos períodos estacionarios, rápidamente progresiva o fulminante. Del tiempo que demore el desarrollo del sistema colateral intra y extracerebral y de su capacidad de compensar los déficit de perfusión existentes, dependerá la evolución, siendo dicho retraso la causa principal de las isquemias recidivantes en el niño. Esto explica la primera fase más grave en la infancia, seguido luego de una etapa de estabilidad y luego una etapa de mayor riesgo de hemorragias en el adulto, como observamos en el primer caso clínico (7).
La evolución de nuestros pacientes fue progresiva en once (los de mayor tiempo de seguimiento), uno de ellos con estabilidad inicial y progresividad en la adolescencia; sin evidencias de progresividad clínica en otros (con una media de seguimiento de dos años) y la mitad fueron revascularizados. La evolución arteriográfica en todos los casos realizados evidenció progresividad en la oclusión del polígono de Willis, lo que confirma la progresividad de la lesión en los vasos involucrados, independientemente de que la cirugía mejore las redes colaterales y la irrigación cerebral en las áreas afectadas.
En nuestra pequeña serie dos niños fallecieron y catorce presentaron discapacidad leve a moderada, en acuerdo con la literatura internacional que expresa que aproximadamente el 50-60% experimentan deterioro gradual de las funciones cognitivas. De ahí la importancia del seguimiento clínico, neuropsicológico y hemodinámico para establecer claramente la evolución; con una mortalidad reportada en niños de 4,3% generalmente en relación a una hemorragia (4,26,27).
Si bien aún no existe consenso sobre la conducta a seguir en la enfermedad de moyamoya asintomática, un reciente estudio prospectivo multicéntrico en Japón sobre 40 pacientes, evidenció que al momento del diagnóstico un 20% tenían infartos por RM y un 40% trastornos en la hemodinamia cerebral. En la evolución seis requirieron cirugía y de los 34 no revascularizados, siete presentaron ACV isquémicos o hemorrágicos (28).
Yamada, evaluando el curso natural de 33 casos asintomáticos, reporta dos muertes por hemorragia y cuatro pacientes con episodios isquémicos a los 44 meses (29). Choi y colaboradores apuntan hacia una mejor calidad de vida en pacientes pediátricos intervenidos mediante técnicas de revascularización (30). Estos informes y nuestra experiencia indican que en estos casos la evaluación y conducta terapéutica no deben diferir entre un grupo y otro, por lo cual la indicación de cirugía se extendería tanto al grupo sintomático como a aquel asintomático con factores de riesgo de isquemia determinados mediante SPECT o PET.
Ningún tratamiento médico ha demostrado eficacia en detener la progresión de la enfermedad, así como tampoco se ha demostrado la utilidad de los calcioantagonistas o inhibidores de la anhidrasa carbónica en evitar la vasorreactividad cerebral a los cambios ácido-base (22). Existe acuerdo en utilizar ácido acetilsalicílico en dosis de 1-5 mg/k/día durante la etapa infantil ante el riesgo de isquemia cerebral en ese rango etario, suspendiendo el tratamiento en edad avanzada por mayor riesgo de hemorragia (7-22).
En cuanto a la cirugía de revascularización existe consenso (en base a trabajos clase 3 y grado de recomendación D), en que actualmente sería la única terapéutica que podría evitar el progreso de la enfermedad, estando indicada fundamentalmente en niños con eventos isquémicos recurrentes o progresivos, deterioro neuropsicológico o con reducción de la perfusión cerebral (4,5,11,13,30). En estos pacientes es de gran valor la realización del PET o SPECT basal/acetazolamida para establecer la hipoperfusión cerebral e identificar a aquellos los niños de mayor riesgo que se beneficiarían de la cirugía, como ya hemos mencionado anteriormente (26,31).
Existen métodos directos e indirectos de revascularización. Los métodos directos consisten en suturar una arteria extracerebral (la arteria temporal superficial generalmente), directamente a una arteria de la superficie cerebral (cerebral media).
Entretanto las técnicas indirectas intentan mejorar el flujo sanguíneo cerebral mediante la colocación de diferentes tejidos vascularizados sobre la superficie cerebral (arteria temporal superficial, músculo, periostio, epiplón).
En nuestra serie siete niños recibieron diez cirugías de revascularización mediante una encefaloduroarteriosinangiosis, sin complicaciones perioperatorias y con buena respuesta clínica y angiográfica. La eficacia clínica se observó en todos los niños operados, con desaparición o atenuación de los síntomas preoperatorios. A nivel angiográfico en los ocho casos con controles posquirúrgicos se objetivó mejoría de la irrigación en el territorio silviano. Robertson y colaboradores encuentran en trece niños, luego de la pialsinangiosis, una clara mejoría clínica y arteriográfica en diez casos, en ausencia de nuevos infartos, pero sin evitar la progresión de su arteriopatía ni el desarrollo de vasos moyamoya (32). Ninguno de nuestros pacientes presentaron isquemias relacionadas con la cirugía, las cuales pueden ocurrir entre un 5-10% de los casos (11,22). Durante la cirugía se procedió a la sutura de la adventicia de la arteria temporal superficial a la piamadre cerebral. En un caso se añadió un trépano frontal con colocación de colgajo de pericráneo sobre la piaaracnoides. Dada la progresividad de la enfermedad, nuestra recomendación actual es realzar cirugía para revascularizar el parénquima cerebral. En una amplia revisión de la literatura publicada de Fung y colaboradores (11) sobre 1.448 revascularizados, en más de 55 publicaciones mundiales, encuentran que 73% recibieron métodos indirectos, el 23% métodos combinados y el resto directos. El 87% evidenciaron mejoría clínica sin diferencias entre los métodos directos e indirectos, con un mayor desarrollo de colaterales con los métodos directos, destacando que en los indirectos las colaterales pueden demorar hasta más de 6 meses en formarse. En cuanto las complicaciones perioperatorias isquémicas (AIT) éstas se observaron en 4,4% a 6,1% de los que recibieron métodos indirectos y combinados respectivamente (11,22). En acuerdo con la gran mayoría de los autores, nuestro equipo quirúrgico prefiere los métodos indirectos (especialmente en menores de 8 años), con el doble fin de aprovechar la capacidad neoangiogénica de los niños y por la dificultad de realizar revascularización directa en vasos de pequeño tamaño. Smith y Scooth en tal sentido han presentado muy buenos resultados con la pyal sinangiosis, que consiste en una encefaloduroarteriosinangiosis modificada con apertura de la piamadre en contacto con la arteria temporal superficial, con el fin de aumentar la angiogénesis por el contacto con LCR a través del factor de crecimiento fibroblástico (4,5).
En los casos hemorrágicos, especialmente en adultos si bien la mayoría de los autores son partidarios de la intervención, asumiendo una mortalidad del 8% anual en los no operados versus un 4% en los operados, aún no está demostrado científicamente que la cirugía de revascularización evite nuevos sangrados. Es por ello que en el año 2004 se inicia un estudio prospectivo multicéntrico, patrocinado por el Comité de Oclusión Espontánea del Polígono de Willis del Ministerio de Salud Japonés, del cual aún no se han comunicado resultados (4,30,33).
Referencias bibliográficas
1. Suzuki J, Takaku A. Cerebrovascular moyamoya disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol 1969; 20: 288-99.
2. Shimizu K, Takeuchi K. Hypoplasia of bilateral internal carotid arteries. Brain Nerve 1957; 9: 37-43.
3. Takeuchi K. Occlusive diseases of the carotid artery: especially on their surgical treatment. Shinkei Kenkyu No Simpo (Tokyo) 1961; 5: 511-43.
4. Quintana Marín L. Enfermedad de moyamoya. J Bras Neurocir 2004; 15(3): 91-104.
5. Smith E, Scooth M. Syndrome moyamoya. Departament of neurosurgery of Children’S Hospital Boston. Rev Argent Neuroc 2005; 19: 31-40.
6. DeVeber G. Arterial ischemic strokes in infants and children: an overview of current approaches. Semin Thromb Hemost 2003; 29: 567-74.
7. Peña-Tapia P, Horn P, Schmiedek. Enfermedad de moyamoya. Rev Neurol 2006; 43(5): 287-94.
8. Takagi Y, Kikuta K, Nozaki K, Hashimoto N. Histological features of middle cerebral arteries from patientes treated for moyamoya disease. Neurol Med Chir (Tokyo) 2007; 47: 1-4.
9. Takagi Y, Kikuta K, Nozaki K, Fujimoto M, Hayashi J, Inamura H, et al. Expresion of hypoxia-inducing Factor 1 alfa and Endoglin in intimal hyperplasia of the middle cerebral artery of patients with moyamoya disease. Neurosurgery 2007; 60(2): 338-45.
10. Pascual-Castroviejo I, López Martín V, Rodríguez Costa T, Tendero A, Palencia R. Síndrome de Nishimoto moyamoya. A propósito de un caso con estudio angiográfico completo. Radiología 1973; 15: 35-42.
11. Fung LW, Thompson D, Ganesan V. Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv Syst 2005; 21: 358-64.
12. González G, Campistol J, Navarro R, Capdevila A, Solá T. Moyamoya en la población infantil. Análisis de una serie occidental y revisión de la bibliografía. Rev Neurol 2008; 46: 385-91.
13. Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (moyamoya disease). Research Committee on Spontaneous Oclussion of the Circle of Willis of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg 1997; 99 (2): 238-40.
14. Houkin K, Nakayama N, Kuroda S, Nonaka T, Shonai T, Yoshimoto T. Novel magnetic resonance angiography stage grading for moyamoya disease. Cerebrovasc Dis 2005; 2: 347-54.
15. Fukuyama I, Umezu R. Clinical and cerebral angiographic evolution of idiopathic progressive occlusive disease of the circle of Willis in children. Brain Dev 1985; (7): 21-37.
16. Bonita R, Beaglehole R. Modification of Rankin Scale. Recovery of motor function after stroke. Stroke 1988; 19(12): 1497-500.
17. Kim SK, Wang KC, Kim IO, Lee DS, Cho BK. Combined encephaloduroarteriosynangiosis an bifrontal encephalogaleoperiostealsynangiosis in pediatric moyamoya disease. Neurosurgery 2002; 50: 88-96.
18. Matsushima Y, Inoue T, Suzuki SO, Fujii K, Fukui M, Hasuo K. Surgical treatment of moyamoya disease in pediatric patients. Comparison between the results of indirect and direct revascularization procedures. Neurosurgery 1992; 31: 401-5.
19. Horn P, Pfister S, Bueltmann E, Vajkoczy, Schmiedek P. Moyamoya-like vasculopathy (moyamoya syndrome) in children. Childs Nerv Syst 2004; 20: 382-91.
20. Ullrich N, Robertson R, Kinnamon D, Scott R, Kieran M,Turner, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology 2007; 68: 932-8.
21. Pascual Castroviejo I, Pascual Pascual S. Epilepsia vascular en la infancia. Rev Neurol 1999; 28: 288-92.
22. Kirton A, deVeber G. Therapeutic Approaches and Advances in Pediatric Stroke. J Am Soc Experiment Neurother 2006; 3: 133-142.
23. Cho DY, Chen TC, Lee HD. Ultra-early decompressive craniectomy for malignant middle cerebral artery infarction. Surg Neurol 2003; 60: 227-32.
24. Schwedt T, Guo Y, Rothner D. “Benign” Imaging Abnormalities in Children and Adolescents with Headache. Headache 2006; 46: 387-98.
25. Seol H, Wang K, Kim S, Hwang Y, Kim K, Cho B. Headache in pediatric moyamoya disease: review of 204 consecutive cases. J Neurosurg (5 Suppl Pediatrics) 2005; 103: 439-42.
26. So Y, Lee H, Kim S, Lee J, Wang K, Cho B, et al. Prediction of the clinical outcome of pediatric moyamoya disease with postoperative basal/acetazolamide stress brain perfusion SPECT after revascularization surgery. Stroke 2005; 36: 1485-89.
27. Punt J. Surgical management of paediatric stroke. Pediatr Radiol 2004; 34: 16-23.
28. Kuroda S, Hashimoto N, Yoshimoto T, Iwasaki Y. Radiological findings, clinical course, and outcome in asymptomatic moyamoya disease. Stroke 2007; 38: 1430-5.
29. Yamada M, Fujii K, Fukui M. Clinical features and outcomes in patients with asymptomatic moyamoya disease-from the results of nation-wide questionnaire survey. No Shinkei Geka 2005; 33: 337-42.
30. Choi J, Kim D, Kim EY, Lee KD. Natural history of moyamoya disease: comparison of activity of daily living in surgery an non surgery groups. Clin Neurol Neurosurg 1997; 99(2): 11-8.
31. Mardinkevicius E, Liutkus D, Gvazdaitis A. Experience of treatment of moyamoya disease at the clinic of Neursurgery of Kaunas University of Medicine. Medicine (Kaunas) 2006; 42: 2.
32. Robertson R, Burrows P, Barnes P, Robson C, Poussaint T, Scott M. Angiographic changes after Pias Synangiosis in Childhood Moyamoya Disease. Am J Neuroradiol; 18: 837-45.
33. Miyamoto S. Study design for a prospective randomized trial of extracranial-intracranial bypass surgery for adults with moyamoya disease and hemorrhagic onset the Japan Adult moyamoya Trial Group. Neurol Med Chir (Tokyo) 2004; 44: 218-9.
Correspondencia: Dr. Gabriel González Rabelino. Cátedra de Neuropediatría
Correo electrónico: viciogon@hotmail.com


{kind=link}
{kind=link}
{kind=link}