










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.79 no.3 Montevideo set. 2008
CASO CLÍNICO
Arch Pediatr Urug 2008; 79(3)
Hiperinsulinismo congénito: revisión de aspectos diagnósticos y terapéuticos a propósito de un caso clínico
Dres. Noelia Speranza 1, Héctor Telechea 1, Gustavo Giachetto 2, María Catalina Pírez 3
1. Residente de Pediatría. Asistente Departamento de Farmacología y Terapéutica.
2. Prof. Agregado Clínica Pediátrica. Prof. Agregado Departamento de Farmacología y Terapéutica.
3. Prof. Clínica Pediátrica. Directora Clínica Pediátrica "A".
Clínica Pediátrica "A", Departamento de Pediatría, Facultad de Medicina, Universidad de la República.
Fecha recibido: 13 de mayo de 2008.
Fecha aprobado: 9 de setiembre de 2008.
Resumen
El hiperinsulinismo congénito (HC) es la causa más común de hipoglicemia persistente en el primer año de vida. Se acompaña de riesgo elevado de daño neurológico irreversible. En los últimos años se ha profundizado en el conocimiento sobre su patogenia, destacándose la heterogenicidad clínica, histológica y genética, con claras implicancias en el diagnóstico y tratamiento. Se describe el caso de un lactante portador de HC que debuta con hipoglicemias sintomáticas a los 5 meses de vida. El objetivo de esta comunicación es, a la luz de los conocimientos actuales, analizar los criterios diagnósticos y terapéuticos de esta patología. Se destaca la importancia de aplicar un algoritmo para guiar el estudio paraclínico y descartar las etiologías más frecuentes asociadas con la hipoglicemia.
Palabras clave:
HIPERINSULINISMO-terapia
HIPOGLICEMIA HIPERINSULINÉMICA
PERSISTENTE DEL LACTANTE
Summary
Congenital hyperinsulinism (HC) is the most common etiology of persistent hypoglycemia in infants. It is associated with a high risk of irreversible neurological damage. Knowledge about its pathogenesis has improved in the past few years. It is characterized by a heterogeneous clinical presentation, histology and genetic which leads to special diagnosis and treatment features.
This is a case report of a 5 month old with HC who had symptomatic hypoglycemia. The objective of this communication is to analyze the diagnosis and treatment criteria of HC. The need to follow a clinical and laboratory guideline for diagnosis will help exclude other causes of hypoglycemia.
Key words:
HYPERINSULINISM-therapy
PERSISTENT HYPERINSULINEMIA
HYPOGLYCEMIA OF INFANCY
Introducción
El hiperinsulinismo congénito (HC) es la causa más común de hipoglicemia persistente en el primer año de vida, con un alto riesgo de daño neurológico irreversible (1-3).
Desde finales de los noventa se han hecho importantes avances en el conocimiento de las bases moleculares de esta enfermedad con claras implicancias en el diagnóstico y el tratamiento (3). Se ha demostrado su heterogenicidad clínica, histológica y genética (1,3,4).
El avance en el conocimiento de estos aspectos ha llevado a cambiar las antiguas denominaciones "hipoglicemia idiopática de la infancia", "hipoglicemia leucina sensible", "insulinoma neonatal", "microadenomatosis pancreática" y "nesidioblastosis" por las actuales "hiperinsulinismo congénito", "hipoglicemia hiperinsulinémica del recién nacido y de la infancia" o "hiperinsulinismo neonatal persistente".
Su incidencia se ha estimado en 1/50.000, pero se describen importantes variaciones según las regiones consideradas (5), entre 1/40.000 en Finlandia a 1/2.500 en áreas de elevada consanguinidad como Arabia Saudita (3,5). En Uruguay se desconoce su frecuencia y al momento de finalizada esta comunicación no se encontraron casos publicados.
El HC se caracteriza por la secreción incontrolada de insulina, expresión de alteraciones en diferentes procesos bioquímicos intracelulares o del transporte iónico a través de la membrana de la célula b pancreática (3). La presencia de hiperinsulinemia impide la movilización de glucosa endógena y explica la hipoglicemia. Éste es el elemento cardinal que guía el diagnóstico.
La hipoglicemia plantea dos problemas. Por un lado el tratamiento sintomático, ya que la hipoglicemia constituye una emergencia médica que puede determinar convulsiones, coma, muerte o secuelas neurológicas irreversibles. Por otro, el reconocimiento de los factores etiológicos que condicionan el tratamiento y pronóstico. El pediatra debe estar familiarizado con este tipo de trastornos y aplicar el razonamiento clínico para guiar los estudios paraclínicos y descartar las etiologías más frecuentes asociadas con hipoglicemia.
A continuación se describe el caso de un lactante portador de HC que debuta con hipoglicemias sintomáticas a los 5 meses de vida. El objetivo de esta comunicación es, a la luz de los conocimientos actuales, analizar los criterios diagnósticos y terapéuticos de esta patología.
Observación clínica
LM, 11 meses, sexo masculino, raza blanca, procedente de zona rural de Canelones. Producto de segunda gestación, embarazo bien controlado, mal tolerado por hipertensión arterial, cesárea de urgencia por preeclampsia a las 38 semanas, peso al nacer 3.710 g (p90), Apgar 7/10. Síndrome de dificultad respiratoria temprana, hipertensión pulmonar, requiere asistencia ventilatoria mecánica durante 5 días y tratamiento con sildenafil. Ictericia neonatal sin conflicto. No convulsiones en el período neonatal, buena evolución posterior. Alimentado con pecho directo exclusivo hasta los 4 meses, incorporación progresiva de otros alimentos a los 6 meses. Buen crecimiento y desarrollo. Certificado esquema de vacunación vigente. Sin antecedentes familiares a destacar, no consanguinidad entre padres.
Ingresa al Centro Hospitalario Pereira Rossell (CHPR) el 1 de junio de 2005, a los 5 meses de edad, por convulsión tónico clónica generalizada, breve, autolimitada, constatándose cifras capilares y venosas de glicemia de 33 mg/dl y 40 mg/dl, respectivamente. Examen físico normal. De la paraclínica realizada durante esa hospitalización se destaca: gasometría venosa pH 7,3, HCO3 24 mEq/l, BE 1,8, pCO2 55 mmHg, glicemia venosa 54 mg/dl, cetonemia negativa, insulinemia 2,2 mU/ml (valor normal entre 2,6-11,1): relación insulinemia/glicemia 0,04 (con ayuno programado de 6 horas); sodio 134 mEq/l, potasio 3,7 mEq/l, calcio 1,15 mmol/l. Dosificación de TSH 3,77 mU/ml (valor normal entre 1,36-8,76); cortisol 17,63 mg/dl (valor normal de los 7 días a 1 año: 4-23); ACTH indosificable (valor normal <46 pg/ml). Ecografía transfontanelar y electroencefalograma (EEG) normales.
Durante la hospitalización se mantiene asintomático. Se otorga alta el 17 de junio de 2005. Se indica alimentación frecuente, cada dos a tres horas. Controlado en Policlínica de Referencia del CHPR se mantiene asintomático con buen crecimiento y desarrollo. No se realizan controles de glicemia.
Reingresa el 12 de diciembre de 2005, a los 11 meses de edad, por nuevo episodio ictal. Dos horas luego de alimentarse, instala bruscamente supraversión ocular con hipotonía generalizada, palidez y sudoración. Consulta inmediatamente, se constata glicemia capilar de 24 mg/dl, se administra suero glucosado vía oral y se traslada al hospital. En el Departamento de Emergencia, 6 horas después, la glicemia capilar era 45 mg/dl.
Del examen físico: peso 9.820 g (p50), talla 75 cm (p50) PC 45 cm (p30). Buen estado general, apirético, frecuencia respiratoria 32 ciclos por minuto, sin tirajes, piel y mucosas normocoloreadas, sin lesiones. Examen neurológico: pares craneanos normales, tono y fuerzas conservadas, sin signos focales, reflejos osteotendinosos normales. Genitales externos normales. Abdomen depresible, sin visceromegalias, fosas lumbares normales. A nivel cardiovascular ritmo regular 100 ciclos por minuto, sin soplos, pulsos presentes normales. Resto del examen físico sin alteraciones.
Durante la hospitalización se somete a ayuno programado de dos horas y se obtienen los siguientes exámenes: gasometría venosa: pH 7,35, HCO3 22 mEq/l, BE –3,7, pCO2 40,7 mmHg; glicemia venosa 34 mg/dl; cetonemia negativa. Insulinemia 15.4 mU/ml; relación insulinemia / glicemia 0,45; péptido C 0,93 ng/ml (valor normal entre 0,51-2,57 ng/ml). Se realiza EEG y ecografía abdominal, que resultan normales. La tomografía axial computada abdominal (TAC) mostró un área hipodensa de 10 mm en el sector distal del cuerpo del páncreas (figuras 1 y 2). La resonancia nuclear magnética (RNM) abdominal fue normal.
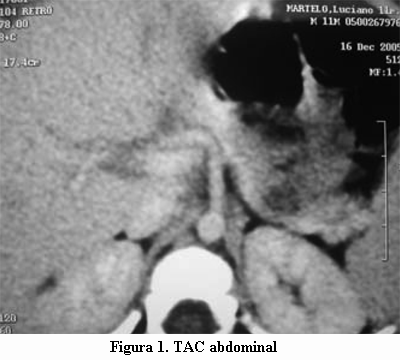
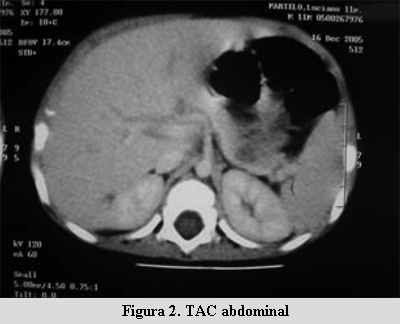
Reitera hipoglicemias asintomáticas en tres oportunidades. Se mantiene con alimentación frecuente y se inicia tratamiento con diazóxido 50 mg vía oral cada 12 horas con monitorización de la glicemia capilar 2 a 4 veces al día.
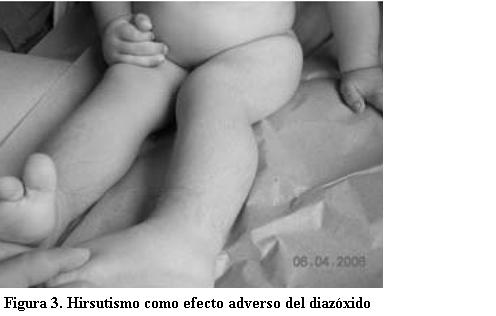
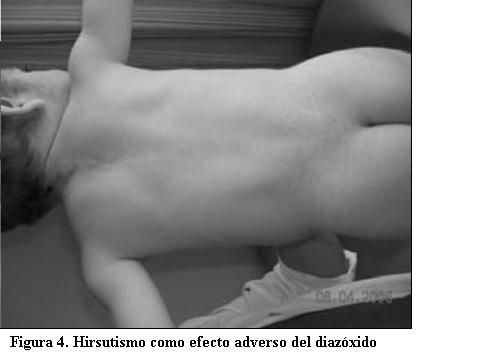
Luego del alta (9 de enero de 2006) fue controlado en la Policlínica de Referencia del CHPR. Se debieron realizar ajustes en la calidad y cantidad de los alimentos y en la dosis de diazóxido administrado hasta lograr, a los 3 meses del alta, adecuado control metabólico. Actualmente recibe 125 mg de diazóxido al día. Presenta hirsutismo como reacción adversa al medicamento (figuras 3 y 4).
Discusión
Este lactante debuta a los 5 meses de edad con una hipoglicemia sintomática sin otras manifestaciones clínicas orientadoras. Ésta constituye una forma de presentación habitual del HC. La hipoglicemia, definida como cifras de glicemia plasmática menores a 40 mg/dl, es el síntoma cardinal que guía el diagnóstico (6-8). En general, los síntomas hipoglicémicos aparecen al reducirse la frecuencia de las ingestas, con ayunos de 4 a 8 horas. Aproximadamente la mitad de los lactantes con HC presentan convulsiones relacionadas con la disminución de la oferta de glucosa a nivel encefálico. Otros síntomas que pueden observarse frecuentemente son: aumento del apetito, decaimiento y excitación. Estos síntomas se relacionan con la activación autonómica y aumento de la descarga adrenérgica en respuesta a la reducción rápida de glucosa. No obstante, es posible que en recién nacidos y lactantes los síntomas sean más sutiles (cianosis, palidez, apnea, hipotermia, hipotonía, mala alimentación, irritabilidad, llanto apagado, letargia) o sean asintomáticos. El examen físico al momento del diagnóstico generalmente es normal como en este paciente. La hepatomegalia leve es frecuente y no excluye el diagnóstico (5).
A pesar que no fue el caso del paciente, es frecuente el antecedente de macrosomía al nacer: 30% de los niños con HC tienen el antecedente de parto por cesárea por dicho motivo.
Frente a un lactante con hipoglicemia sintomática, sin otros elementos clínicos orientadores, es imprescindible seguir un algoritmo de estudios a fin de poder llegar al diagnóstico etiológico (figura 5).
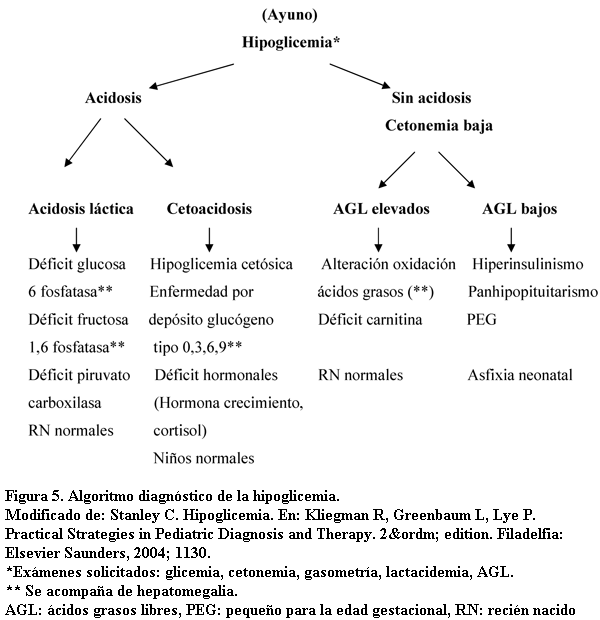
Por frecuencia, la hipoglicemia hipocetonémica, debe orientar a HC. Este fue el planteo inicial en este paciente. La confirmación del diagnóstico no se logró durante la primera internación. Los datos orientadores más importantes hasta ese momento eran la ausencia de acidosis, la cetonemia negativa y el valor de insulinemia hallado para los valores de hipoglicemia observados. Este último aspecto es muy importante ya que normalmente las hipoglicemias marcadas no se acompañan de insulinemias normales. Las concentraciones de insulina que son inadecuadamente elevadas en presencia de hipoglicemia sugieren un defecto en la secreción basal (1,7).
La ausencia de cetosis es uno de los primeros elementos a considerar en este diagnóstico. Fuera del período neonatal, el nivel de cuerpos cetónicos guarda relación con el grado de hipoglicemia del paciente. Ante la presencia de hipoglicemia sería esperable cetonuria francamente positiva (tres o cuatro cruces en la tira de orina). La ausencia de cetonemia o cetonuria sugiere hiperinsulinismo o un defecto de la oxidación de los ácidos grasos (cetogénesis inapropiada por trastorno de la beta oxidación que se diagnostica con la dosificación concomitantemente los niveles de ácidos grasos libres y de beta hidroxibutirato). En ayuno el cociente entre estos dos parámetros es menor a 3; un cociente mayor sugiere una cetogénesis inapropiada (8). El HC se acompaña de cetonemia negativa o baja y dosificación de ácidos grasos libres y de beta hidroxibutirato en plasma bajos. En la mayoría de las otras causas de hipoglicemia, excepto la galactosemia y la intolerancia a la fructosa, se observan cetonuria y cetonemia.
Otros aspectos clínicos y paraclínicos orientadores durante la primera internación fueron la ausencia de visceromegalias, frecuentemente observadas en las enfermedades por depósito de glucógeno o errores innatos del metabolismo; la adecuada tolerancia a los alimentos que básicamente descarta la galactosemia; el crecimiento, examen físico y valores hormonales de cortisol, ACTH y TSH normales que alejaban la posibilidad de panhipopituitarismo o déficit específico de cortisol.
Para confirmar el diagnóstico es imprescindible extraer las muestras de sangre al momento que se constata la hipoglicemia, ya que la mayoría de estos exámenes pierden valor si no se analizan en este contexto.
Sin embargo no siempre es posible objetivar la hipoglicemia, por lo que, como ocurrió en este paciente, es necesario provocarla con un ayuno programado. En general esta prueba puede requerir hasta 8 a 12 horas de ayuno. Debe realizarse con el paciente hospitalizado bajo estricta monitorización y se recomienda realizarlo temprano en la mañana para contar con todo el personal de salud para su evaluación, conociendo previamente las condiciones de extracción y almacenamiento de cada muestra. Es recomendable extraer muestras plasmáticas y urinarias extra de modo de poder utilizarlas para estudios que puedan necesitarse en la evolución (5 ml de plasma o suero y 10 ml de orina) (6,8) (tabla 1). Hay que recordar que en estos casos valores de glicemia de 50 mg/dl son suficientes para el diagnóstico de hipoglicemia.
Durante la segunda internación, a las 2 horas de haber comenzado el ayuno, se obtuvieron valores de glicemia plasmática de 0,34 mg/dl y pudo obtenerse muestras para el resto de los estudios. Así fue posible confirmar el diagnóstico de HC por la presencia de glicemia plasmática menor a 40 mg/dl o 50 mg/dl con ayuno provocado; insulinemia mayor a 10 mU/ml; y relación insulinemia/glicemia mayor o igual a 0,4-0,5 mU/ml/mg/dl (7,8).
En general se observa aumento del péptido C concomitantemente a la hipoglicemia. Esta sustancia, que no tiene una función biológica conocida, se segrega junto a la insulina y es un representante útil de la secreción endógena total de ésta, no se halla elevada en las hipoglicemias secundarias a la administración exógena de insulina, como puede verse en el síndrome de Munchhausen por poder o en pacientes diabéticos (7). Sin embargo en los lactantes portadores de HC puede observarse niveles de insulina y péptido C normales durante los periodos de hipoglicemia (5).
En algunos casos puede ser necesario realizar otros estudios orientadores de HC como la estimulación con glucagón. En estos pacientes, una vez que se constata hipoglicemia (menor a 50 mg/dl), al administrar glucagón se produce un aumento mayor a 30-40 mg/dl de la glicemia, lo que traduce que la movilización de glucosa se ha visto limitada por la insulina y que los mecanismos de glucogenólisis están intactos (7).
Se distinguen dos forma anatomopatológicas de la enfermedad, focal y difusa. La forma focal representa aproximadamente el 40% de los casos y difiere claramente del adenoma pancreático del adulto (5). Generalmente son alteraciones nodulares entre 2,5 y 7,5 mm, la mayoría se ubican en la cabeza del páncreas. La diferenciación de estas dos formas es importante ya que tanto el tratamiento como el pronóstico varían sustancialmente. Mientras que en las lesiones focales la resección quirúrgica local generalmente es curativa, en las difusas (que no responden al tratamiento médico) se requiere una pancreatectomía subtotal, con alto riesgo de aparición de diabetes (3). Los métodos diagnósticos de elección utilizados actualmente para diferenciar estas formas son:
1. Estimulación selectiva de la arteria pancreática con calcio, midiendo la respuesta de insulina en muestras obtenidas en la vena hepática.
2. Sampling venoso pancreático, obteniéndose muestras selectivas de la cabeza, cuerpo y cola del páncreas para determinar los niveles de insulina, glicemia y péptido C (3,9,10).
Para algunos autores, el sampling venoso es la técnica más precisa y segura para localizar las formas focales en niños (10,11). Los pacientes con alteraciones focales presentarán niveles de insulina y péptido C elevados en una o varias muestras contiguas y bajo en el resto de los sitios. Los portadores de lesiones difusas tienen valores elevados en todas las muestras. Ninguno de estos estudios fue posible realizar en este paciente, si bien la respuesta al tratamiento médico ha sido hasta el momento satisfactoria.
La tomografía con emisión de positrones con F fluoro L dopa es una técnica imagenológica en desarrollo que podría ofrecer ventajas frente al sampling venoso (12).
En contraste con el hiperinsulinismo congénito focal, los adenomas pancreáticos pueden ser detectados por estudios imagenológicos como TAC, RNM, centellograma con octeótrido o ecografía transduodenal, si bien el diagnóstico es angiográfico. Estas técnicas imagenológicas no son útiles para identificar las lesiones focales del HC. Esto explica la ineficacia de estos métodos para el diagnóstico, sumado a la excepcionalidad de aquel diagnóstico en pediatría (5,8). En este paciente se realizaron TAC y RNM abdominales a fin de evaluar morfológicamente el páncreas, pero los hallazgos de la TAC no se confirmaron en la RNM y dado que no fueron concluyentes no se tuvieron en cuenta para el diagnóstico.
También se utilizan técnicas histológicas intraoperatorias que ayudan a completar el diagnóstico y precisar la extensión de la resección quirúrgica (4,5).
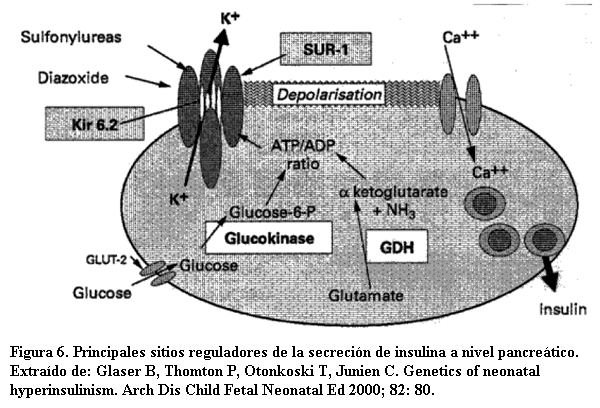
En aproximadamente 50% de los casos se han descrito diversos tipos de defectos genéticos en la secreción de insulina: mutaciones de genes que codifican para las subunidades SUR 1 y KIR6.2 del canal de potasio ATP dependiente (al cerrarse estos canales provocan la despolarización celular y la activación de canales de calcio voltaje dependientes), y mutaciones para las enzimas glutamato deshidrogenasa, glucoquinasa o L- 3hidroxiacil deshidrogenasa de cadena corta que determinan aumentos de la relación ADP/ATP necesaria para el cierre de los canales de potasio (figura 6) (3,7,12). Las formas graves de inicio neonatal suelen correlacionarse con alteraciones del canal de potasio y las formas leves con alteraciones enzimáticas. También desde el punto de vista molecular pueden distinguirse las formas difusas y focales. Mientras las primeras expresan alteraciones de la línea germinal que afectan a todas las células, y por tanto todo el páncreas, las formas focales son producto de la alteración heredada del alelo paterno SUR o KIR6.2 junto a la pérdida espontánea de material genético materno del cromosoma 11p15.1 restringida a un foco de células pancreáticas (12). Estos conocimientos se están incorporando al diagnóstico genético de esta enfermedad.
El tratamiento del HC comprende la prevención y tratamiento de la hipoglicemia y el tratamiento específico del hiperinsulinismo, en el que se centrará la discusión.
En este paciente se realizó un tratamiento en base a alimentación frecuente, con carbohidratos de lenta metabolización y uso de diazóxido. El tratamiento farmacológico de elección incluye a este fármaco (5), para algunos autores combinado con otros fármacos como la hidroclorotiazida (13,15,16). El diazóxido es un agonista de los canales de potasio ATP dependiente, que hiperpolariza las células beta pancreáticas e inhibe la secreción de insulina (17). La dosis varia entre 5 a 25 mg/kg/día vía oral (generalmente 15 mg/kg/día), administrado de 2 a 4 veces al día. Se inicia con dosis entre 5 a 10 mg/kg/día con aumentos semanales de a 5 mg/kg/día según la respuesta clínica y paraclínica. El efecto hiperglicemiante generalmente comienza una hora luego de administrado y dura más de 8 horas (5,17-19). La eficacia del diazóxido se define como la normalización de los niveles de glucosa (>54 mg/dl) medido antes y después de cada comida en pacientes que se están alimentando normalmente, luego de un ayuno nocturno fisiológico y de haber detenido el uso de glucosa intravenosa o cualquier otra medicación por cinco días consecutivos (5). La respuesta clínica completa a este fármaco es variable, entre 15% y más de 60% (15). Esta diferencia puede reflejar la variabilidad de pacientes seleccionados para tratamiento médico, la eficacia de diferentes dosis y la conocida heterogenicidad genética e histopatológica asociada a esta enfermedad (16). Se considera "resistencia al diazóxido" cuando el paciente presenta al menos un valor de glicemia < 54mg/dl con las condiciones arriba mencionadas (2,5).
Generalmente es un fármaco bien tolerado. El hirsutismo es el efecto adverso más frecuente, como se observó en este paciente. Probablemente se relaciona a un aumento de hormonas androgénicas. También pueden evidenciarse hipotensión arterial, edema, alteraciones electrolíticas, alopecia, cambios en la voz, anorexia, disgeusia, cefalea, hiperuricemia leve, eosinofilia, trombocitopenia y síntomas extrapiramidales. Las reacciones de hipersensibilidad se manifiestan como rush, leucopenia y fiebre (18). La monitorización del tratamiento con este fármaco a largo plazo incluye controles periódicos de la presión arterial, hemograma, evaluación del crecimiento y desarrollo incluyendo maduración ósea y psicológica. En un estudio de seguimiento prospectivo de 22 niños que recibieron diazóxido, tres desarrollaron hirsutismo, dos presentaron retención de líquidos y ninguno presentó alteraciones del crecimiento (12).
La hidroclorotiazida actúa sinérgicamente con el diazóxido en la apertura de los canales de potasio. El uso combinado con diazóxido disminuye la retención hidrosalina y potencia el efecto hiperglicemiante. Se administra a una dosis de 7 a 10 mg/kg/día vía oral cada 12 horas (15,16). Se han utilizado otros fármacos como el octeótrido, un análogo de la somatostatina reservado para pacientes que no responden al diazóxido como alternativa al tratamiento quirúrgico; calcioantagonistas (nifedipina) aunque su eficacia no ha sido aun claramente demostrada (5,15,16), y glucocorticoides. El uso de estos últimos a largo plazo es controvertido debido a su inadecuado perfil de seguridad y tolerabilidad (15,16).
Los pacientes que requieren tratamiento médico, en general se mantienen dependientes del tratamiento farmacológico, si bien algunos presentan buena respuesta y pueden alcanzar una remisión completa relativamente rápido, en menos de 16 meses en las formas focales y de 60 meses en las formas difusas. Esta remisión puede asociarse a una apoptosis de las células beta, por lo que algunos autores recomiendan tratamiento conservador aun en pacientes con resistencia al tratamiento médico. Esta posibilidad de remisión justifica la recomendación de suspender transitoriamente, una vez al año, bajo estricta supervisión médica el tratamiento farmacológico.
Las indicaciones para el tratamiento quirúrgico incluyen resistencia o intolerancia al tratamiento médico (5) y cuando se halla diagnosticado adecuadamente una forma focal (15). Se considera resistencia al tratamiento cuando a pesar de alimentación fraccionada y tratamiento farmacológico adecuado se mantenga dependencia a la administración de glucosa enteral o parenteral para mantener glicemias sobre 60 mg/dl (13,15).
El tratamiento quirúrgico de las formas focales generalmente es curativo (11,20,21). En los portadores de formas difusas, está indicada la pancreatectomía subtotal (95% del órgano). Estas resecciones amplias se asocian con riesgo de hipoglicemia recurrente o persistente, diabetes mellitus (95% de los casos) o intolerancia a la glucosa. Se requiere una evaluación anual de la secreción residual de insulina, glicemia e insulinemia pre y posprandiales, los niveles de hemoglobina glicosilada (A1c) y test de tolerancia oral a la glucosa (5). En un estudio prospectivo que evaluó 22 niños de los cuales 16 debieron someterse a pancreatectomía subtotal, cuatro desarrollaron intolerancia a la glucosa, y tres de ellos evolucionaron a diabetes mellitus (12).
La aparición de hipoglicemia desde el nacimiento y la necesidad de tratamiento quirúrgico parecen constituir dos de los factores pronósticos más importantes para la aparición de complicaciones neurológicas por hipoglicemia. En un estudio retrospectivo que valoró la evolución de 90 pacientes con diagnóstico de hiperinsulinismo se evidenció retardo psicomotor severo en siete de los niños, alteración psicomotora moderada en 12 y epilepsia en 16 (2). En otra serie de 26 pacientes, 10 con formas clínicas graves debieron ser intervenidos quirúrgicamente por falta de respuesta al tratamiento médico y siete presentaron secuelas neurológicas. En el resto de los niños tratados con diazóxido, sólo uno presentó secuelas neurológicas (23).
En este paciente se destaca la importancia de la aplicación de un algoritmo de estudios paraclínicos basado en algunos elementos epidemiológicos y clínicos orientadores para poder llegar al diagnóstico de HC y la importancia del seguimiento evolutivo para poder determinar la respuesta terapéutica.
Referencias bibliográficas
1. Stanley C. Hyperinsulinism in infants and children. Pediatr Clin North Am 1997; 44: 363-74.
2. Menni F, de Lonlay P, Sevin C, Touati G, Peigne C, Barbier V, et al. Neurologic outcomes of 90 neonates and infants with persistent hyperinsuinemic hypoglycemia. Pediatrics 2001; 107(3): 476-9.
3. Glaser B, Thornton P, Otonkoski T, Junien C. Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed 2000; 82: 79-86.
4. Sempoux C, Guiot Y, Lefevre A, Fékété N, Jaubert F, Saudubray J, et al. Neonatal hyperinsulinemic hypoglycemia: heterogeneity of the syndrome and keys for differential diagnosis. J Clin Endocrinol Metabol 1998; 83(5): 1455-61.
5. De Lonlay P. Persistent hyperinsulinemic hypoglycemia. Orphanet encyclopedia [en línea]. Paris: Orphanet, 2003 <http://www.orpha.net/data/patho/GB/uk-PHHI.pdf > [consulta: 25 ene 2008]
6. Stanley C. Hypoglycemia. En: Kliegman R, Greenbaum L, Lye P. Practical Strategies in Pediatric Diagnosis and Therapy. 2 ed. Philadelphia: Elsevier Saunders, 2004: 1121-31.
7. Sperling M. Hipoglicemia. En: Behrman R, Kilegman R, Jenson H. Nelson. Tratado de pediatría. 16 ed. México: McGraw-Hill Interamericana, 2001: 481-94.
8. Camberos M, Abdenur J, Cresto J. Hipoglicemia. En: Pombo M. Tratado de Endocrinología Pediátrica. Madrid: Mc Graw Hill-Interamericana, 2002: 1102-21.
9. Stanley C, Thornton P, Ganguly A, Macmullen C, Underwood P, Bhatia P, et al. Preoperative Evaluation of Infants with Focal or Diffuse Congenital Hyperinsulinism by Intravenous Acute Insulin Response Tests and Selective Pancreatic Arterial Calcium Stimulation. J Clin Endocrinol Metab 2004; 89(1): 288–96.
10. Dubois J, Brunelle F, Touati G, Scbag G, Nuttin C, Thach T, et al. Hyperinsulinism in children: diagnostic value of pancreatic venous sampling correlated with clinical, pathological and surgical outcome in 25 cases. Pediatr Radiol 1995; 25: 512-6.
11. Brunelle F, Negre V, Barth M, Fékété C, Czernichow P, Saudubray J, et al. Pancreatic venous samplings in infants and children with primary hyperinsulinism. Pediatr Radiol 1989; 19: 100-3.
12. Guerrero-Fernández J, González I, Espinoza L, García R. Hiperinsulinismo congénito. Revisión de 22 casos. An Pediatr (Barc) 2006; 65(1): 22-31.
13. De Lonlay P, Poggi-Travert F, Fournet J, Sempoux C, Vici C, Brunelle F, et al. Clinical features of 52 neonates with hyperinsulinism. NEJM 1999; 340(15): 1169-75.
14. Sempoux C, Guiot Y, Rahier J. The focal form of persistent hyperinsulinemic hipoglycemia of infancy. Diabetes 2001; 50: S182-S183.
15. Aynsley-Green A, Hussain K, Hall J, Saudubray J, Fékété C, de Lonlay P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed 2000; 82: F98-107.
16. Dunne M, Cosgrove K, Shepherd R, Aynsley-Green A, Lindley K. Hyperinsulinism in Infancy: From Basic Science to Clinical Disease. Physiol Rev 2004; 84: 239-75.
17. Hardman J, Limbird L. Molinoff P, Ruddon R, Goodman Gilman A. Goodman & Gilman. Las bases farmacológicas de la terapéutica. 9 ed. México: Mc Graw-Hill Interamericana, 1996.
18. Martindale W. The Complete Drug Reference. 34 ed. London: The Pharmaceutical Press, 2005.
19. Hernández I, Hodgson M, Cattani A. Hiperinsulinismo neonatal persistente. Análisis diagnóstico de dos casos clínicos. Rev Med Chile 2004; 132: 995-1000.
20. Fékété C, de Lonlay P, Jaubert F, Rahier J, Brunelle F, Saudubray J. The Surgical Management of Congenital Hyperinsulinemic Hypoglycemia in Infancy. J Pediatr Surg 2004; 39: 267-9.
21. Adzick S, Thornton P, Stanley C, Kaye R, Ruchelli E. A Multidisciplinary Approach to the Focal Form of Congenital Hyperinsulinism Leads to Successful Treatment by Partial Pancreatectomy. J Pediatr Surg 2004; 39: 270-5.
22. Cherian M, Abduljabbar M. Persistent hyperinsulinemic hypoglycemia of infancy: long term outcome following 95% pancreatectomy. J Pediatr Endocrinol Metab 2005; 18(12): 1441-8.
23. Cresto J, Abdenur J, Bregada I, Martino R. Long term follow up of persistent hyperinsulinaemic hypoglycemia of infancy. Arch Dis Child 1998; 79:440-4.
Correspondencia: Dra. Noelia Speranza.
Bv. Artigas 1550, 3er piso. Montevideo, Uruguay
E mail: noeliasperanza@gmail.com


{kind=link}