










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.79 no.1 Montevideo 2008
PAUTAS
Arch Pediatr Urug 2008; 79(1)
Protocolo diagnóstico y terapéutico del síndrome de Guillain-Barré
Dres. Rosario Taboada 1, Gabriel González 2, Alicia García 3, Marta Alberti 4, Cristina Scavone 5
1. Pediatra. Ex. Asistente de Clínica Pediátrica. Postgrado de Unidad de Cuidados Intensivos del Niño. Facultad de Medicina UdelaR.
2. Neuropediatra. Profesor Agregado de Neuropediatría. Facultad de Medicina UdelaR.
3. Pediatra Intensivista. Profesor Adjunto de Unidad de Cuidados Intensivos del Niño. Ex Profesor Adjunto de Pediatría. Facultad de Medicina UdelaR.
4. Pediatra Intensivista Profesor de Unidad de Cuidados Intensivos del Niño. Facultad de Medicina UdelaR.
5. Neuropediatra. Profesor de Neuropediatría. Facultad de Medicina UdelaR.
Criterios diagnósticos
(Asbury et al, 1978-1990)
1. Pilar clínico.
2. Estudio del LCR.
3. Estudio electrofiosiológico.
1. Hallazgos clínicos
- Debilidad muscular progresiva de más de una extremidad. La debilidad muscular se desarrolla rápidamente (gravedad máxima a las cuatro semanas).
- Ausencia de reflejos de estiramiento.
- Simetría relativa.
- Signos y síntomas sensitivos relativamente leves.
- Pares craneanos: VII par afectado en 50% (< 5% puede iniciarse por pares craneanos.)
- La recuperación comienza 2 a 4 semanas después de haber alcanzado la gravedad máxima.
- Alteraciones autonómicas.
- Sin fiebre al comienzo de los síntomas neurológicos.
2. Hallazgos en el LCR
- Disociación albúmino-citológica.
- Proteínas elevadas después de la primera semana.
- Células: no más de 10 leucocitos por mm3 (hay autores que aceptan hasta 50 leucocitos / mm3).
3. Hallazgos neurofisiológicos
- Velocidad de conducción disminuida en 60% (no uniforme por característica segmentaria del proceso).
- Bloqueo de conducción en 80%.
- Aumento de latencias distales.
- Latencia de onda F aumentada o ausente
Inconvenientes en el diagnóstico
Los criterios diagnósticos del síndrome de Guillain-Barré (SGB) no abarcan el espectro clínico completo de este trastorno. Entre 10% y 20% de los pacientes no cumplen estos criterios, son las variantes de SGB. Las variantes clínicas incluyen:
- Fiebre inicial.
- Pérdida sensitiva grave.
- Progresión luego de cuatro semanas o recaída leve.
- Cese de la progresión sin recuperación o con secuelas permanentes importantes.
- Alteración de esfínteres.
- Afectación del sistema nervioso central (SNC).
Estas formas variantes propuestas por Romper se dividen en:
- Formas regionales.
- Síndrome de Fisher.
- Cérvico-faringo-braquial (frecuentemente ptosis).
- Parálisis oculofaríngea.
- Predominio paraparesia.
- Parálisis facial bilateral con parestesias distales.
- Oftalmoplejia con Ac. GQ1b.
- Formas funcionales.
- Ataxia sin disartria o nistagmo.
- Forma sensitiva pura.
- Pandisautonómica.
- Forma motora pura.
- Forma axonal.
Por lo tanto, el diagnóstico se debe basar en hallazgos clínicos, de laboratorio y electrofisiológicos consistentes y en la exclusión de otras condiciones con formas de presentación similar.
Clasificación de gravedad del SGB
En función de la clínica; Hughes los clasifica en escalas funcionales:
1. Síntomas y signos leves, pero que le permiten desempeñar actividades de andar, correr con dificultad, vestirse, comer y aseo.
2. Puede caminar más de 5 metros sin ayuda, pero no saltar, correr o realizar actividades de cuidado personal.
3. Puede caminar más de 5 metros pero con ayuda.
4. Está en cama.
5. Necesita ventilación asistida.
6. Muerte.
45% ingresan en estadio 3 o menor, 40% en estadio 4 y 15% en estadio 5.
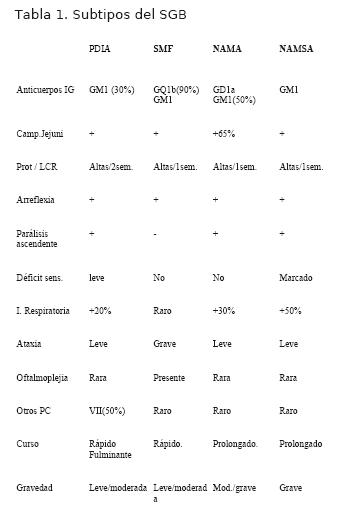
Subtipos del SGB (tabla 1)
- Polineuropatía inflamatoria desmielinizante aguda (PDIA).
- Síndrome de Miller Fisher (SMF).
- Neuropatía axonal motora aguda (NAMA).
- Polineuropatía axonal sensitivo-motora aguda (NAMSA).
Rasgos dudosos para el diagnóstico (criterios Asbury-Ropper)
- Presencia de un nivel sensitivo.
- Marcada o persistente asimetría de los síntomas o de los signos.
- Disfunción esfinteriana persistente y grave.
- Más de 50 células/mm3 en el líquido cefalorraquídeo.
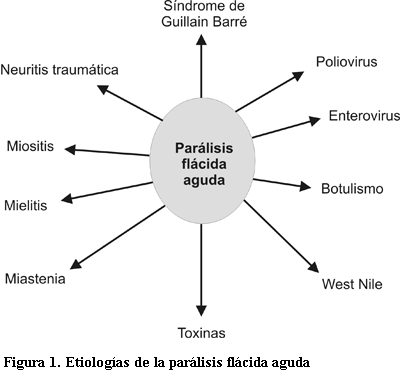
Diagnósticos diferenciales de parálisis fláccida en la infancia
Es una definición operativa, enmarcada en la campaña de la Organización Mundial de la Salud (OMS), de la década de 1980, con el objetivo de erradicar la polio del mundo, por lo que intenta incluir todos los casos de probable poliomielitis.
Esta definición incluye tres grandes aspectos:
1. Instalación aguda o hiperaguda en menos de cinco días.
2. Cuadro clínico caracterizado por una disminución o pérdida de fuerzas y tono muscular (flacidez) de una o más extremidades (espinal) pudiendo acompañarse de participación craneal.
3. Presentarse en niños menores de 15 años.
Esta definición abarca fundamentalmente las enfermedades del sistema nervioso periférico, que cursan con paresia e hipotonía y en las cuales se debe descartar obligatoriamente el virus de la polio, aunque como veremos existen múltiples etiologías (figura 1, tabla 2).
Rasgos que excluyen el diagnóstico
- Diagnóstico de:
- botulismo;
- miastenia grave;
- poliomielitis;
- neuropatía tóxica.
- Trastornos en el metabolismo de las porfirinas.
- Difteria reciente.
- Síndrome sensitivo puro sin debilidad.
Tratamiento
- Ingreso hospitalario.
- Declaración obligatoria, parálisis fláccida. (Sanidad).
Manejo de soporte
Adecuado monitoreo respiratorio, hemodinámico, nutricional, metabólico, y de la debilidad motora.
Criterios de ingreso a unidad de cuidado intensivo pediátrico (UCIP)
Se recomienda en:
- Compromiso del ritmo: bloqueo o bradicardia.
- Rápida progresión de debilidad
- Infección (sepsis, neumonía)
- Taquiarritmias
- Indicación de monitoreo continuo
- Complicaciones: trombosis venosa profunda, tromboembolismo pulmonar, infarto agudo del miocardio)
- Aérea (insuficiencia respiratoria)
- Labilidad hemodinámica
- Otras indicaciones incluyen:
- Síndrome de Miller Fisher.
- Variantes clínicas con compromiso de nervios craneales.
Manejo del dolor
El dolor es frecuente (65%-85%). Se presenta a nivel lumbar y de miembros inferiores.
- Frecuentes cambios de posición.
- Analgésicos comunes: acetaminofén o antiinflamatorios no esteroideos.
- En ocasiones se requiere empleo de narcóticos.
- Antineuríticos: gabapentina a 15 mg/kg/día dividido en tres dosis (nivel de evidencia II, recomendación grado B).
- Amitriptilina como coadyuvante en el manejo del dolor.
Manejo respiratorio
El SGB puede evolucionar rápidamente a falla respiratoria de tipo hipodinámico.
En las etapas iniciales la saturometría puede ser normal.
La función respiratoria puede verse comprometida sin signos clínicos de insuficiencia ventilatoria.
La debilidad muscular impide utilizar los músculos accesorios respiratorios.
El manejo de la función respiratoria incluye:
- Permeabilidad de las vías aéreas.
- Capacidad del paciente para toser y expectorar.
- Evaluación de la mecánica ventilatoria.
- Habilidad para deglutir.
- Signos de hipoxemia e hipercapnia.
Los criterios para iniciar asistencia ventilatoria mecánica son:
- Gasometría arterial que muestre hipoxemia y/o hipercapnia (no esperar a que tenga trabajo respiratorio para realizarla)
- Capacidad vital menor a 10 ml/kg.
- Parálisis bulbar o disfagia (o debilidad en la lengua) con peligro de broncoaspiración.
El empleo de traqueostomía debe limitarse a pacientes en quienes se requiera ventilación mecánica prolongada.
Manejo del compromiso disautonómico
La taquicardia sinusal no suele requerir tratamiento.
El manejo de la respuesta disautonómica: beta bloqueadores tipo propranolol (1 mg/kg/día).
La hipotensión no responde a inotrópicos, debe ser manejada con volumen (sin nivel de evidencia).
Las bradiarritmias y la asistolía son los trastornos del ritmo mas peligrosos; tener cuidado con estímulos vagotónicos (succión, intubación, sonda nasogástrica, etcétera).
Manejo nutricional
El paciente con SGB inicialmente tiene un estado hipercatabólico secundario al estrés; requiere aportes elevados de proteínas y calorías, por tanto la dieta debe ser hiperprotéica e hipercalórica: las necesidades básicas energéticas se estiman de acuerdo a peso, estatura y edad.
La vía enteral es de primera elección para el soporte nutricional (mantiene la integridad y el papel inmunológico del intestino, profilaxis de úlcera por estrés, bajo costo, menor riesgo de complicaciones (sin nivel de evidencia).
En pacientes con trastorno de la deglución, la nutrición debe ser enteral a través de sondas flexibles. La administración será en infusión por gastroclisis.
El paciente debe estar en posición antirreflujo.
Una vez superado el trastorno deglutorio, se inicia la vía oral, previa valoración por fonoaudiología para corroborar la deglución adecuada.
Se hará protección de gastritis y/o de úlcera de stress con ranitidina 1.5 mg/kg dosis cada 6 horas máximo 50 mg.
Apoyo psicológico y psiquiátrico
El soporte emocional al paciente y su familia hace parte del tratamiento. Es recomendable una educación temprana para el paciente y sus familiares referente a la enfermedad.
Los antidepresivos pueden utilizarse como adyuvante para el manejo de los trastornos del sueño, dolor y consecuencias emocionales. (Sin nivel de evidencia).
Manejo específico: Inmunoterapia
Las inmunoglobulinas intravenosas y la plasmaféresis son útiles, en etapas tempranas.
Indicaciones:
- Empeoramiento en la situación funcional.
- Estadio inicial 3 (imposibilidad de caminar en forma independiente).
- Compromiso bulbar.
En las formas leves, capaces de caminar, no hay consenso si deben o no ser tratadas con inmunoterapia, hay autores que sugieren que sería innecesario si en la segunda semana no pierden la deambulación.
Entre ambas terapias (inmunoglobulinas, plasmaféresis) los últimos ensayos clínicos, realizados en Europa y Norteamérica, no muestran diferencias significativas, no estando indicado su empleo combinado.
Contraindicaciones absolutas de la inmunoterapia:
- Deficiencia selectiva de IgA.
- Anafilaxia luego de iniciar la infusión de IgG intravenosa.
Contraindicaciones relativas:
- Falla renal.
- Insuficiencia cardíaca congestiva.
Dosis: inmunoglobulinas por vía intravenosa: 400 mg/Kg/día durante cinco días, o 2 g/kg en un día (a elegir). En niños se prefiere el uso de inmunoglobulinas por su tolerancia, en planes de 2 g/kg en cinco o dos días.
Las recaídas en niños son raras, y en estos casos se recomienda repetir el mismo tratamiento.
Los corticosteroides
Actualmente no están indicados, si bien fueron controvertidos, y en los últimos años el grupo holandés de Van Koningsveld postuló que existe un efecto positivo sinérgico con la inmunoglobulina intravenosa, aunque los resultados no mostraron diferencias significativas.
Fisioterapia
Objetivos:
1. Atención a la correcta posición y alineación de miembros y cabeza para evitar la aparición de retracciones o distensiones músculo-tendinosas y ligamentarias.
2. El tratamiento postural será de vital importancia en el desarrollo futuro de muchas patologías, siendo a veces necesario el uso de ortesis funcionales o de posicionamiento (antirrotatorios).
3. Cuidado de la piel, evitando la aparición de úlceras por presión.
4. Prevención de complicaciones respiratorias (atelectasia, neumonías por disminución de la ventilación pulmonar).
5. Mantenimiento de las amplitudes fisiológicas de movimiento.
6. Prevención de problemas circulatorios, principalmente por éstasis venoso.
7. Estimulación sensorial.
Evolución y seguimiento
- Mediante clasificación funcional de Hughes; evaluar la respuesta al tratamiento y pronóstico.
- Repetir el estudio eléctrico a las 3 semanas, y en función de la evolución a los 6 y 12 meses.
- Estudio serológico en sangre y LCR (sangre: segunda determinación a las tres semanas, si se realizan estudios de anticuerpos angigangliósidos).
- El egreso hospitalario se recomienda una vez alcance una escala de Hughes de 3.
- Se recomienda iniciar tempranamente la terapia física.
- Continuar controles periódicos ambulatorios de neuropediatría y rehabilitación.
Pronóstico
La evolución del SGB en la infancia es mejor que en adultos, la mortalidad alcanza el 1%-5% de los niños, de los que un 25% quedan con secuelas en general leves en forma de debilidad distal de miembros inferiores.
La mayor afectación clínica es un signo de peor pronóstico. Las formas axonales en general tienen un peor pronóstico que las formas desmielinizantes.
Referencias bibliográficas
1. Cerisola A, Capote G, Scavone C. Síndrome de Guillain Barré en pediatría. Diferentes formas de presentación y dificultades en el diagnóstico precoz. Rev Neurol 2007; 44(12): 725-32.
2. Monteiro JP, Fonseca S, Proenca J. Síndrome de Guillain Barré en edad pediátrica. Experiencia de laUnidad de Neuropediatría de un hospital portugués. Neurology 2006; 42(3): 144-9.
3. Tellería A, Calzada DJ. Síndrome de Guillain Barré. Neurology 2002; 34(10): 966-76.
4. González G. Parálisis flácida en la infancia. Arch Pediatr Urug 2006; 77(3): 308-12.
5. Pérez A, Frignla J. Síndrome de Guillain Barré. Bol Pediatr 2006; 46(supl 1): 49-55.
6. Williams CR. Síndrome de Guillain Barré. Rev Med Hondur 2006 ; 35 : 230-6.
7. Schttlender JG, Lombardi D, Toledo A. Compromiso respiratorio en el sindrome de Guillain Barré. Medicina (Buenos Aires) 1999; 59: 705-9.
8. Puga MS, Padrón A, Bravo R. Sindrome de Guillain Barré. Rev Cuba Mil 2003; 32(2): 137-42.
Correspondencia: Dra. Alicia García
Lorenzo Pérez 3171/501. CP 11300.
Montevideo. Uruguay.
Correo electrónico: agape@montevideo.com.uy


{kind=link}