










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.78 no.3 Montevideo Sept. 2007
CASO CLÍNICO
Arch Pediatr Urug 2007; 78(3)
Síndrome de Rett: microcefalia con retardo mental y autismo. Reporte de un caso
Dres. Jorge Arturo Aviña Fierro 1, Daniel Alejandro Hernández Aviña 2
1. Dismorfología Pediátrica, UMAE. Centro Médico Nacional de Occidente. IMSSS, Guadalajara, México.2. Urgencias Médicoquirurgicas, Cruz Verde. Servicios Médicos Municipales. Guadalajara, México.
Fecha de recibido: 5 de julio de 2007.
Fecha de aprobado: 24 de agosto de 2007.
Resumen
El síndrome de Rett es un grave proceso del desarrollo neurológico que afecta exclusivamente mujeres, se considera una enfermedad monogénica dominante ligada al cromosoma X debida a mutaciones en el gen MECP2 codificador de la proteína 2 de unión a metil-CpG. La paciente fue normal hasta los 18 meses de vida cuando inició con deterioro en sus destrezas psicomotoras: marcha atáxica y pérdida del movimiento intencionado de las manos; alteración social y de conducta con autismo infantil y retardo mental importante. Esta enfermedad suele, frecuentemente, estar mal diagnosticada como autismo o parálisis cerebral y carece de tratamiento específico.
Palabras clave:
SÍNDROME DE RETT
MICROCEFALIA
RETRASO MENTAL
TRASTORNO AUTÍSTICO
Summary
Rett syndrome is a severe neurodevelopment disorder which affects exclusively women. It is considered a dominant disease linked to the X chromosome; it is due to mutations in the MECP2 gene which encodes the methyl-CpG binding 2 protein. This patient had a normal development until 18 months of age, then a deterioration of her psychomotor skills with ataxia and loss of purposeful use of the hands began; the behavioral and social areas were also affected with autism and progression to profound mental retardation. The syndrome is misdiagnosed often as autism or cerebral palsy and has no specific treatment.
Key words:
RETT SYNDROME
MICROCEPHALY
MENTAL RETARDATION
AUTISTIC DISORDER
Introducción
El síndrome de Rett es una alteración neurológica progresiva observada solamente en mujeres, descrita inicialmente en 1966 por Rett (1). En 1983, la enfermedad recibió mayor difusión, por una serie de 35 casos realizada por Hagberg (2). Las pacientes reportadas mundialmente son aproximadamente 2.000, con una incidencia de una paciente por cada 15.000 nacidas vivas (3). Suele diagnosticarse en forma errada como autismo o parálisis cerebral (4); el diagnóstico de síndrome de Rett se realiza a través de la valoración clínica, basándose en los siguientes criterios modificados en 1996 por Clarke: desarrollo prenatal y perinatal normales, pérdida de habilidades previamente adquiridas como lenguaje y comunicación, perímetro cefálico normal al nacimiento y luego microcefalia adquirida, importante retardo en el desarrollo, pérdida de habilidades de las manos, hallazgos autistas, marcha atáxica y movimientos estereotipados de las manos (5). Otros criterios accesorios son: convulsiones, anormalidades electroencefalográficas, apnea, hiperventilación, disfunción respiratoria, escoliosis, retardo del crecimiento, pies fríos y espasticidad con desgaste muscular (6). No existen marcadores bioquímicos o genéticos para diagnosticar el síndrome (7).
Caso clínico
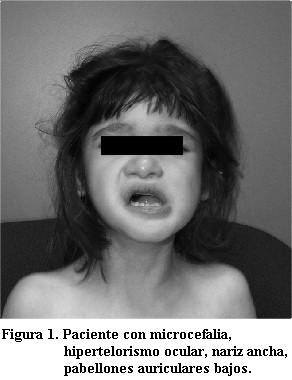
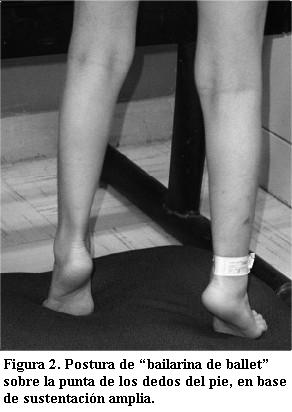
Paciente femenina, de 5 años de edad, producto de embarazo normal a término, parto sin complicaciones, llanto y respiración espontáneas; peso al nacer 3.300 g, talla 50 cm, perímetro cefálico 35 cm, sin malformaciones detectables; alimentación inicial al seno materno, a los 5 meses fórmula maternizada y ablactación. Padre 32 años, madre 29, matrimonio no consanguíneo; dos hermanos varones 7 y 6 años, todos sanos. El primer año y medio de vida con crecimiento y desarrollo normales, a los 18 meses de edad mostró dificultad para deglutir, crisis de irritabilidad y frecuentes períodos de agitación, luego indiferencia a los estímulos externos, torpeza en el uso de las manos y caídas frecuentes; llevada a consulta se concluyó deterioro neurológico por parálisis cerebral infantil de causa desconocida, no requirió medicación. Posteriormente la paciente evolucionó a retardo mental importante hacia los dos años de edad, con disminución de la movilidad espontánea y confinamiento a la cuna, padeciendo complicaciones respiratorias frecuentes y hospitalizaciones por neumonías. En el último ingreso se notaron datos de autismo y dismorfia craneofacial con microcefalia (perímetro cefálico de 45 cm), hipertelorismo ocular, nariz ancha, pabellones auriculares bajos, filtrum plano y epicanto (figura 1). Paciente caquéctica con retraso psicomotor y retardo mental, autista en llanto constante, sin lenguaje, sólo emitiendo gruñidos, espástica con moderada escoliosis, sacudidas torácicas espontáneas, incoordinación de movilidad de extremidades, movimientos estereotipados de manos en frote continuo; en la posición erecta adoptaba postura de “bailarina de ballet” parándose sobre la punta de los dedos del pie, en base de sustentación amplia (figura 2).
Discusión
La causa de la enfermedad permanece desconocida, como sólo afecta mujeres se asocia con el cromosoma X en una enfermedad dominante, que sería letal en los hombres (8). El 95% de los casos son esporádicos por mutación de novo; un pequeño porcentaje se debe a que alguno de los progenitores tiene mosaicismo somático o de línea germinal, y existe un pequeño número de casos de recurrencia familiar que tienen como región génica de interés la Xq28 (9). Las mutaciones suelen presentarse en el gen MECP2 que produce la proteína de unión 2 de metil-CpG, causando su alteración una pérdida de función de factores neurotróficos por disrupción en metilación de unión a ADN y alteración en represores transcripcionales (10). Los estudios neuropatológicos han revelado hipofunción en sinapsis de neuronas aminérgicas del tallo cerebral e hipersensibilidad del receptor; existiendo una ruptura de los circuitos neuronales con anormalidades de incremento en glutamato e inhibición del ácido gamma-aminobutírico en receptores sinápticos; finalmente se disminuye del tamaño de algunas neuronas, su ramificado dendrítico y el número de sus prolongaciones espinosas. Las zonas más afectadas son los lóbulos frontales, el núcleo caudado y el mesencéfalo (11,12).
La aparente normalidad temprana del desarrollo neurológico ha sido siempre uno de los criterios de la enfermedad (13), pero se ha reportado en una revisión de videos de pacientes con la enfermedad, que la valoración cuidadosa detectaba movimientos generales anormales, postura rígida, protusión de lengua, asimetría en la apertura y cierre de los ojos, estereotipias de las manos, movimientos anormales de los dedos, brotes de expresiones faciales anormales, sonrisa bizarra y temblor; concluyéndose que las alteraciones son más tempranas pero pasan desapercibidas (14). El diagnóstico diferencial se realiza con autismo, parálisis cerebral atáxica, síndrome de Angelman y alfa talasemia recesiva con retardo mental ligado a X. No hay tratamiento específico, sólo medidas de sostén y fisioterapia; algunas pacientes que mantienen movilidad, se benefician al convivir con caballos (equinoterapia) o pueden realizar actividades físicas en agua (hidroterapia); si la movilización de la paciente es casi nula puede intentarse musicoterapia (15).
La paciente se encuentra actualmente en manejo multidisciplinario para control nutricional y de su progresión ponderal, fisioterapia para mejorar tonicidad corporal, vigilancia pediátrica para evitar complicaciones respiratorias y se le efectúa hidroterapia en tina acuática. La familia no requirió consejo genético por paridad satisfecha y tener método de planificación familiar definitivo.
Referencias bibliográficas
1. Rett A. Uber ein zerebral-atrophisches Syndrome bei Hyperammonemie. (On an unusual brain atrophy syndrome with hyperammonemia in childhood). Wien Med Wochenschr 1966; 116: 723-6.
2. Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol 1983; 14: 471-9.
3. Hagberg B, Hagberg G. Rett syndrome: epidemiology and geographical variability. Eur Child Adolesc Psychiatry 1997; 6: 5-7.
4. Calderón R, Gramajo O, Sevilla R, Carrera JP, Peña F, Bolaños G. Síndrome de Rett: una causa frecuente, poco reconocida de retraso mental. Rev Mex Pediatr 1989; 56: 191-200.
5. Clarke A. Rett syndrome. J Med Genet 1996; 33: 693-9.
6. Nieto BM, Nieto JM, Siljeström ML. Clinical phenotypes of classic Rett syndrome. Rev Neurol 2003; 36 Suppl 1: S146-52.
7. Coronel Carvajal C. Síndrome de Rett: un nuevo reto para los pediatras. Rev Cuba Pediatr 2002; 74: 162-7.
8. Akbarian S, Jianq Y, Laforet G. The molecular pathology of Rett syndrome: synopsis and update. Neuromolecular Med 2006; 8: 485-94.
9. Evans JC, Archer HL, Whatley SD, Clarke A. Germiline mosaicism for a MECP2 mutation in a man with two Rett daughters. Clin Genet 2006; 70: 336-8.
10. Williamson SL, Christodoulou J. Rett syndrome: new clinical and molecular insights. Eur J Hum Genet 2006; 14: 896-903.
11. Johnston MV, Blue ME, Naidu S. Rett syndrome and neuronal development. J Child Neurol 2005; 20: 759-63.
12. Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol 2005; 20: 747-53.
13. Nomura Y, Segawa M. Natural history of Rett syndrome. J Child Neurol 2005; 20: 764-8.
14. Einspieler C, Kerr AM, Prechtl HF. Is the early development of girls with Rett dirsorder really normal? Pediatr Res 2005; 57: 696-700.
15. Lotan MN, Ben-Zeev B. Rett syndrome. A review with emphasis on clinical characteristics and intervention. Scientific World Journal 2006; 6: 1517-41.
Correspondencia: Dr. Jorge Arturo Aviña Fierro.
Alberto Cossío 1432, Huentitán El Alto.
Guadalajara 44390. México.
Correo electrónico: avinafie@megared.net.mx

