










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.78 no.2 Montevideo June 2007
CASO CLÍNICO
Arch Pediatr Urug 2007; 78(2)
Síndrome de trisomía 9p: características clínico-evolutivas y citogenéticas.
Seguimiento de doce años
Dra. Alicia Vaglio 1, Bach. Búrix Mechoso 2, PhD. Andrea Quadrelli 3, Dr. Roberto Quadrelli 4
1. Subdirectora del Instituto de Genética Médica, Hospital Italiano.2. Candidato a Magíster en Ciencias, PEDECIBA.
3. Lic. en Biología. Magíster en Antropología Social. Doctora en Antropología Social. Universidad Federal de Rio Grande del Sur.
4. Director del Instituto de Genética Médica. Académico Titular de la Academia Nacional de Medicina.
Instituto de Genética Médica, Hospital Italiano, Montevideo.
Fecha recibido: 30 de mayo de 2007
Fecha aprobado: 10 de julio de 2007
Resumen
La trisomía 9p es una anomalía cromosómica que se define por la duplicación parcial o completa del brazo corto de un integrante del par cromosómico 9. Clínicamente se caracteriza por retraso mental y psicomotor, malformaciones craneofaciales distintivas y anomalías de manos y pies. En el presente trabajo describimos el seguimiento clínico durante 12 años de una niña con diagnóstico de trisomía del brazo corto del cromosoma 9, con un cariotipo no balanceado definido por la siguiente fórmula: 46,XX,t(9;21)(q10;q10),+i(9)(p10). Los hallazgos fenotípicos observados en la niña ilustran las deficiencias asociadas con una duplicación completa del brazo corto del cromosoma 9 y ayudan en el asesoramiento genético para esta particular anomalía cromosómica.
Palabras clave:
TRISOMÍA
CROMOSOMAS HUMANOS PAR 9
ABERRACIONES CROMOSÓMICAS
TRASTORNOS DE LOS CROMOSOMAS
Summary
Trisomy 9p is a chromosomal anomaly defined by partial or complete duplication of the short arm of one of the members of the 9 pair chromosome. Clinical findings include growth and mental retardation, characteristic craniofacial malformations and hand-foot anomalies. We report a 12 year follow-up of a female patient with trisomy 9p with an unbalanced karyotype defined as: 46,XX,t(9;21)(q10;q10),+i(9)(p10). The observed phenotypic findings illustrate the deficiencies associated with a complete duplication of the short arm of chromosome 9 and can aid in the genetic counseling of this particular chromosomal anomaly.
Key words:
TRISOMY
CHROMOSOMES, HUMAN, PAIR 9
CHROMOSOME ABERRATIONS
CHROMOSOME DISORDERS
Introducción
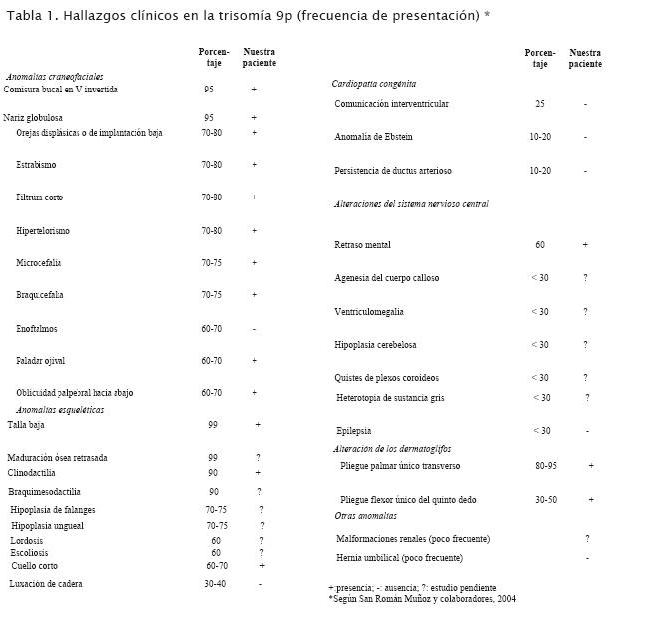
La trisomía 9p, o síndrome de Rethoré, es una anomalía cromosómica que se define por la duplicación parcial o completa del brazo corto del cromosoma 9. El primer caso fue descrito por Rethoré y colaboradores en 1970 (1). El patrón de malformaciones fue descrito por Centerwall y Beatty-DeSana en 1975 (2). Desde entonces se han reportado cerca de 150 pacientes con trisomía 9p completa o parcial (3). En la mayoría de los casos la trisomía 9p es resultado de una translocación cromosómica entre el cromosoma 9 y un segundo autosoma presente en uno de los padres; en una proporción menor es resultado de un cambio genético espontáneo (de novo) que ocurre por razones desconocidas en una etapa temprana en el desarrollo embrionario (3,4). La variabilidad fenotípica es grande debido al tamaño variable del fragmento trisómico involucrado y a la correspondiente monosomía o trisomía resultante. Los hechos clínicos incluyen retraso mental y psicomotor, orejas displásicas o de implantación baja, hipertelorismo, nariz globulosa o prominente, comisura bucal en V invertida y anomalías de las manos y pies (2,3,5-8). En la tabla 1 se presentan algunos de los hallazgos clínicos más frecuentes (9).
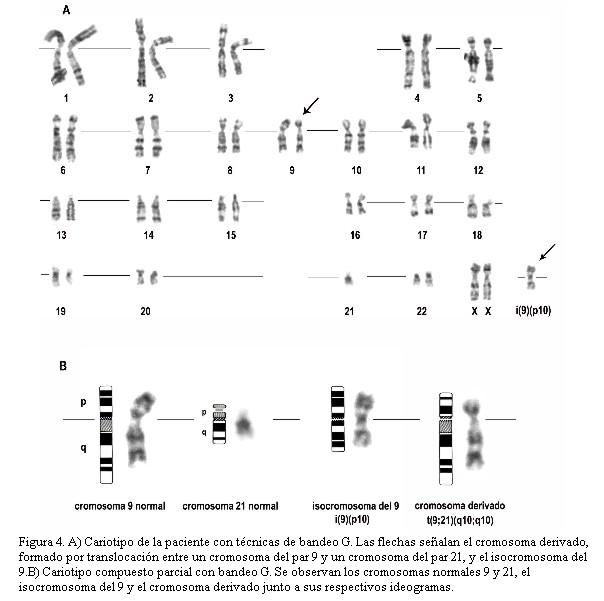
La trisomía 9p representa la cuarta trisomía autosómica más frecuente después de las trisomías 21, 13 y 18 (3,9). Sin embargo, hasta donde sabemos, no se ha publicado ningún caso en la literatura médica uruguaya. En el presente trabajo describimos el prolongado seguimiento clínico (12 años) de una niña con diagnóstico de trisomía del brazo corto del cromosoma 9 con un cariotipo no balanceado definido por la siguiente fórmula: 46,XX,t(9;21)(q10;q10),+i(9)(p10). Esta fórmula establece la presencia de 46 cromosomas, incluyendo el par de cromosomas sexuales XX, define la presencia de una translocación entre los cromosomas 9 y 21, la presencia de un isocromosoma del brazo corto (p) del 9 y la ausencia de un cromosoma 21 libre.
La paciente presenta características clínicas distintivas relacionadas con una trisomía 9p. Como plantea San Román Muñoz y colaboradores (9), el interés de estos casos radica en la importancia de orientar el diagnóstico para el asesoramiento genético y su tratamiento temprano. La publicación de casos adicionales y la evaluación continua permiten un mejor diagnóstico y manejo del paciente.
Material y método
Paciente
Se trata de una niña nacida a las 36 semanas de edad gestacional por parto espontáneo asistido con fórceps, resultado del primer embarazo de madre adolescente. No se reportaron exposiciones tóxicas o eventos inusuales durante la gestación. La madre tenía retardo mental y una edad de 14 años al momento del nacimiento. Padre desconocido. El peso al nacer fue de 2,4 kgs (percentil 3), talla de 46 cms (percentil 3) y perímetro cefálico de 32 cms (percentil 3). La puntuación de Apgar fue de 9 al minuto y 10 a los 5 minutos.
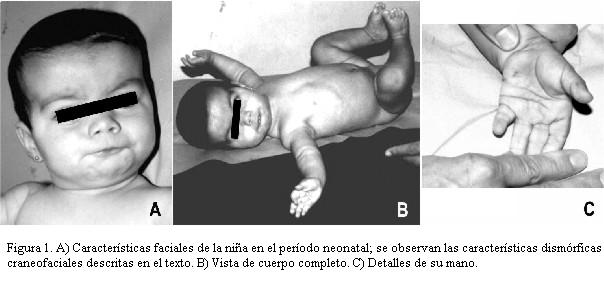
Se examinó por primera vez en el Instituto de Genética Médica a los 2 meses y 20 días de edad. Su peso fue de 3,8 kg (percentil 3), talla de 51,5 cm (percentil < 3) y perímetro cefálico de 36 cm (percentil 3). En la observación clínica se destacaron características craneofaciales dismórficas, incluyendo microbraquicefalia, hipertelorismo ocular, estrabismo convergente, hendiduras palpebrales “antidown”, comisura bucal en V invertida, nariz prominente, orejas de implantación baja, filtrum corto, cuello corto y paladar ojival (figura 1). También se observó braquimesodactilia y clinodactilia del quinto dedo bilateral con pliegue palmar único transverso. Hipotónica.
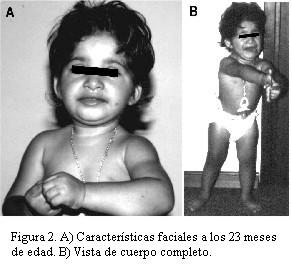
Se examinó nuevamente a los 23 meses de edad. Su peso fue de 8,5 kg (percentil < 3), talla de 75 cm (percentil < 3) y perímetro cefálico de 44 cm (< 2 DS). Presentó parámetros de crecimiento y desarrollo significativamente retrasados. Comenzó a gatear al año. Se paró sola a los 21 meses. Al momento del examen clínico, se mantenía parada pero no caminaba sola. Sostén cefálico a los 3 meses y medio. Sedestación con apoyo a los 6 meses y medio. Alimentación con semilíquidos. Alergia a leche de vaca. Ausencia de lenguaje. Se mantienen las características clínicas ya descritas en el primer examen (figura 2).
La paciente se reevaluó clínicamente a los 7 años y 6 meses de edad. Su peso fue de 21 kg (percentil 25), talla de 98 cm (percentil < 3) y perímetro cefálico de 47,5 cm (< 2 DS). Marcha liberada a los 3 años y medio. Control esfinteriano diurno desde los 5 años. Sin control esfinteriano nocturno. Sin pérdidas dentarias. Escasas palabras sueltas.
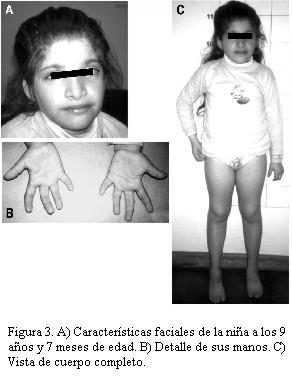
Una nueva evaluación clínica realizada a los 9 años y 7 meses de edad mostró un peso de 23 kg (percentil < 3), talla de 113,5 cm (percentil < 3) y un perímetro cefálico de 48,7 cm (< 2 DS). Control esfinteriano completo a los 8 años. Presenta dificultades para alimentarse. Usa mamadera de noche. Abdomen globuloso. Cumple órdenes en forma parcial. Se viste con ayuda. Vocabulario muy escaso, con palabras sueltas. No toma medicación psiquiátrica. En tratamiento con antialérgicos (figura 3).
A los 12 años, pesó 30 kg (percentil 3) y midió 130 cm (percentil <3). Ingiere pocos volúmenes repartidos en varias ingestas. Habla con palabras cortadas. No está escolarizada. Lee algunas letras. Se viste sin ayuda. No está medicada. Le molesta el ruido y los lugares con mucha gente. Muy poca interacción social y retardo mental evidente.
Análisis citogenético
Los análisis cromosómicos se realizaron en linfocitos de sangre periférica de la paciente y de su madre. Se aplicaron técnicas de bandeo G usando protocolos estándar. Se analizaron un total de 20 metafases con el análisis completo de cuatro cariotipos.
Resultados
La figura 4A muestra el cariotipo con técnicas de bandeo G de la paciente. En las metafases analizadas se observó un total de 46 cromosomas incluyendo el par de cromosomas sexuales (XX). En lugar del par de cromosomas 9 normales se observó un cromosoma 9 normal y un cromosoma derivado resultado de la translocación entre un cromosoma del par 9 y un cromosoma del par 21 (figura 4B). Asimismo, se observó la presencia de un isocromosoma del brazo corto (p) del 9 [i(9)]. El i(9) es un cromosoma anormal compuesto por dos brazos idénticos (p y q) al brazo corto del cromosoma 9 (un brazo duplicado), con ausencia del brazo largo (figura 1B). La paciente presenta una trisomía para todo el brazo corto del cromosoma 9 definida por el siguiente cariotipo: 46,XX,t(9;21)(q10;q10),+i(9)(p10). Los análisis cromosómicos de la madre mostraron un cariotipo 46, XX normal. No fue posible realizar estudio citogenético del padre.
Discusión
Las características clínicas más comunes de la trisomía 9p, incluyendo parámetros de crecimiento y desarrollo retrasados, retardo mental, características faciales típicas como nariz prominente, orejas de implantación baja, cuello corto y clinodactilia, fueron todas observadas en nuestra paciente (tabla 1). El seguimiento clínico durante 12 años de la niña, tal cual se presenta en este trabajo, hace conocer y amplía la literatura disponible del cuadro clínico de los pacientes con trisomía 9p (3), especialmente en nuestro medio.
El diagnóstico de este desorden se basa en el estudio cromosómico de células obtenidas a partir de una muestra de sangre periférica mediante técnicas de Giemsa, bandas GTG, e hibridación in situ fluorescente (FISH), complementadas por la hibridación genómica comparada (CGH), que permite determinar la región del cromosoma 9 implicada y su tamaño (9-11). Para el caso presentado, los estudios de FISH y CGH (no realizados aún) permitirían una caracterización molecular de la anomalía cromosómica observada, si bien no son necesarios para la confirmación del diagnóstico de síndrome de trisomía 9p. Por otra parte, las exploraciones complementarias básicas recomendables para definir la expresión fenotípica son: estudio óseo radiológico (que confirme hipoplasia de falanges), electrocardiograma y ecocardiograma (generalmente normales), ecografía abdominal, pruebas de neuroimagen y valoración del desarrollo psicomotor (9).
El pronóstico de los pacientes con trisomía 9p es muy variable. El grado de retraso psicomotor, la existencia de cardiopatía o la aparición de crisis epilépticas son los factores más determinantes (9).
No hay una explicación definida de por qué la trisomía 9p es la cuarta anomalía cromosómica más frecuente en recién nacidos después de la trisomía 21, la trisomía 18 y la trisomía 13. Una explicación posible se relaciona con el hecho de que todos estos cromosomas (9, 13, 18 y 21) tienen relativamente pocos genes, y la elevada frecuencia de estas anomalías en la progenie puede reflejar un desequilibrio genómico relativamente bajo y, por lo tanto, una mayor tasa de supervivencia (9,12). También la alta frecuencia de trisomía 9p puede deberse a la existencia de puntos de ruptura cromosómicos especialmente frágiles en una o más regiones del brazo corto del cromosoma 9 (3).
A modo de conclusión, el caso presentado aquí destaca la presencia de rasgos clínicos definidos en la trisomía parcial 9p proporcionando información que optimiza el asesoramiento genético y el pronóstico asociado con esta particular anomalía cromosómica.
Referencias bibliográficas
1. Rethoré MO, Larget-Piet L, Abonyi D, Boeswillwald M, Berger R, Carpentier S, et al. Sur quatre cas de trisomie pour le bras court du chromosome 9. Individualisation d’une nouvelle entité morbide. Ann Genet 1970; 13(4): 217-32.
2. Centerwall WR, Beatty-DeSana JW. The trisomy 9p syndrome. Pediatrics 1975; 56(5): 748-55.
3. Littooij AS, Hochstenback R, Sinke RJ, van Tintelen P, Giltay JC. Two cases with partial trisomy 9p: molecular cytogenetic characterization and clinical follow-up. Am J Med Genet 2002; 109(2): 125-32.
4. Van Ravenswaaij-Arts C, Van der Looij E, Smeets D. Trisomy 9p: a clinical Picture and the importance of examining the family. Ned Tijdschr Geneeskd 1999; 143(13): 682-6.
5. Sutherland GR, Carter RF, Morris LL. Partial and complete trisomy 9: delineation of a trisomy 9 syndrome. Hum Genet 1976; 32(2): 133-40.
6. Young RS, Reed T, Hodes ME, Palmer CG. The dermatoglyphic and clinical features of the 9p trisomy and partial 9p monosomy syndromes. Hum Genet 1982; 62(1): 31-9.
7. Wilson GN, Raj A, Baker D. The phenotypic and cytogenetic spectrum of partial trisomy 9. Am J Med Genet 1985; 20(2): 277-82.
8. Smart RD, Viljoen DL, Fraser B. Partial trisomy 9 – further delineation of the phenotype. Am J Med Genet 1988; 31(4): 947-51.
9. San Román Muñoz M, Herranz Fernández JL, Tejerina Puente R, Arteaga Manjón-Cabeza R, López Grondona F. Trisomía 9p. An Pediatr (Barc) 2004; 61(4): 336-9.
10. de Pater JM, Ippel PF, van Dam WM, Loneus WH, Engelen JJ. Characterization of partial trisomy 9p due to insertional translocation by chromosomal (micro) FISH. Clin Genet 2002; 62(6): 482-7.
11. Teebi AS, Gibson L, McGrath J, Meyn MS, Breg WR, Yang-Feng TL. Molecular and cytogenetic characterization of 9p abnormalities. Am J Med Genet 1993; 46(3): 288-92.
12. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science 2001; 291(5507): 1304-51.
Correspondencia: Dra. Alicia Vaglio.
Instituto de Genética Médica, Hospital Italiano. Bulevar Artigas 1632, ZP 11600. Montevideo, Uruguay.
Correo electrónico: rquadr@dedicado.net.uy


{kind=link}
{kind=link}
{kind=link}