










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.78 no.2 Montevideo jun. 2007
CASO CLÍNICO
Arch Pediatr Urug 2007; 78(2)
Neuro-Behçet: a propósito de un caso clínico
Dres. Gabriel González, Rosana Pérez, Rosario Satriano, Ma. Teresa Rotondo
Dirección Nacional de Sanidad Policial. Hospital Policial “Insp. Gral. Uruguay Genta”. Departamento de Neuropediatría y Psiquiatría.Fecha de recibido: 6 de noviembre de 2007.
Fecha aprobado: 17 de julio de 2007.
Resumen
Se describe el caso de un varón de 7 años que se presenta clínicamente con episodios recidivantes de fiebre, aftas y manifestaciones de meningoencefalitis, con hiperproteinorraquia y pleocitosis linfocitaria. Se descartaron otros diagnósticos diferenciales. Con imágenes patológicas en RNM de iso o hipointensidad en T1, hiperintensidad en T2 y Flair, en mesencéfalo, pedúnculos cerebelosos, protuberancia, cuerpo calloso, ganglios basales y cápsula interna y hemisferios cerebrales. En la evolución agrega signos cutáneos cumpliendo con los criterios diagnósticos establecido Grupo Internacional de Estudio de Enfermedad de Behçet. Se realizó tratamiento corticoideo e inmunosupresor.
Destacamos que los criterios diagnósticos internacionales tienen un valor relativo en la etapa prepuberal, dado que las manifestaciones oculares y genitales son infrecuentes y el compromiso parenquimatoso neurológico puede ser su forma de expresión clínica principal.
Palabras clave:
SíNDROME DE BEHÇET-diagnóstico
SíNDROME DE BEHÇET-terapia
VASCULITIS
DIAGNóSTICO DIFERENCIAL
Summary
The case of a 7-year- old Uruguayan boy with recurrent episodes of fever, oral aphthas lesions and meningoencephalitis with elevated protein levels and lymphocytic pleocytosis is presented. Other diagnoses were excluded. MRI showed in acute stages CNS involvement in iso- or hipointense in T1- weighted images and hyperintense in T2-weighted or fluid- attenuated inversion recovery (FLAIR) images in mesodiencephalic junction, cerebellar peduncles, pons, corpus callosum, basal ganglia and internal capsule, and cerebral hemispheres. In the evolution skin lesions appeared, fulfilling the International Study Group Criteria for the Diagnosis of Behçet' Disease. Corticosteroids and immunosuppressant treatment was done.
The main difficulty in children is having all the criteria for diagnosis; especially when recurrent genital ulceration and eye lesion rarely appear at this age.
Key words:
SYNDROME-diagnosis
BEHÇET SYNDROME-therapy
VASCULITIS
DIAGNOSIS, DIFFERENTIAL
Introducción
La enfermedad de Behçet es un trastorno inflamatorio crónico, multisistémico, ocasionado por una vasculitis de origen desconocido. La primera descripción data del siglo V a. C., cuando Hipócrates describió un síndrome caracterizado por coroiditis recurrente, asociado a úlceras orales y genitales. En 1930 el oftalmólogo griego Benedictus Adamantiadis reporta un caso similar con la tríada característica de úlceras orales, genitales y uveítis, agregando compromiso articular. En 1937 Hulusi Behçet describe la enfermedad que hoy lleva su nombre (1); planteando como hipótesis que la misma pudiera ser inducida por virus (2). A las manifestaciones multisistémicas ya mencionadas, se asocian alteraciones del sistema nervioso, cardiovascular, pulmonar y gastrointestinal. Si bien su distribución es mundial, existen amplias diferencias geográficas, siendo la mayor prevalencia en lo que se ha denominado la ruta de la seda, o sea, países del mediterráneo oriental y Lejano Oriente (3). El compromiso del sistema nervioso es una de las manifestaciones mas graves de la enfermedad, la cual se observa en el 5 y 25% de los pacientes (2,4-10). La excepcionalidad de esta enfermedad en nuestro medio y su presentación en la edad pediátrica motivan esta comunicación.
Caso clínico
Escolar de 7 años, de sexo masculino, sin antecedentes familiares a destacar, procedente de medio suburbano. Producto de embarazo y parto normal peso: 3.560 g. Talla: 50 cm. Perímetro craneano: 36 cm, sin enfermedad perinatal. Con buen crecimiento y desarrollo.
Enfermedad actual
Comienza a los 5 años con episodios recidivantes de aftas orales y fiebre. Internado se diagnostica en la primera oportunidad angina de Vincent con hemograma con 10.550 glóbulos bancos, hemoglobina 12 g/dl, plaquetas 224.000 elementos/mm3, velocidad de eritrosedimentación de 100 mm en la primera hora, estudios bacteriológicos en orina y sangre negativos; cultivo faríngeo bacilos Gram positivos fusiformes, espirilos; tratado con penicilina por vía intravenosa por 21 días.
Ingresa en dos oportunidades observándose mal aspecto general, fiebre persistente, aftas orales, realizándose estudios bacteriológicos y TORCH negativos, siendo dado de alta con diagnóstico de síndrome febril prolongado de origen desconocido.
Ingresa a los 6 años con fiebre, aftas orales, síndrome meníngeo y signos focales neurológicos caracterizados por: afectación de pares craneanos, III par incompleto a derecha (ptosis y estrabismo divergente), VII par periférico a izquierda, signo de liberación piramidal a derecha y ataxia cerebelosa. Estudio citoquímico de líquido cefalorraquídeo (LCR): líquido claro, Pandy +, proteínas 0,71 g/dL, glucorraquia 0,40 g/dL, GB: 70 elementos/mm3 con 70% de mononucleares. Cultivo bacteriológico: estéril para hongos y bacilo tuberculoso. Virológico: PCR negativa para: familia herpes virus, enterovirus y arbovirus. Anticuerpos antiborrelia negativos en LCR y plasma. HIV negativo.
Proteinograma electroforético: no se realizó.
Inmunoelectroforesis en LCR: no presenta bandas oligoclonales en zona de gammaglobulinas.
Poblaciones linfocitarias: 14% de linfocitos, CD3 745 elementos/mm3 (56,8%), CD4 313 elementos/mm3 (23,9%), CD8 275 elementos/mm3 (21%). Índice CD4/CD8: 1,1. CD19: 243 elementos/mm3 (18%), CD16 273 elementos/mm3 (20,8%).
En suma: aumento de células NK y linfocitos T doble negativo (CD3 +, CD4 y CD8 negativos).
Células LE, ANCA, inhibidor lúdico, anticuerpos anticardiolipina y anticuerpos antinucleares negativos.
Funcionalidad renal y hepática normal.
EEG: sufrimiento cerebral difuso.
RNM: áreas hiperintensas en la secuencia T2 y Flair en: hemisferio cerebeloso izquierdo subcortical, tronco encefálico a nivel protuberancial y de pedúnculos cerebrales, en región subtalámica bilateral y gangliobasal izquierdo, lóbulo parietal subcortical y paraventricular derecho (figuras 1 y 2).
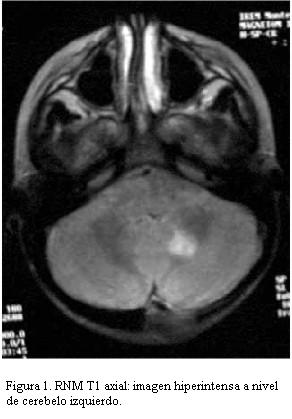
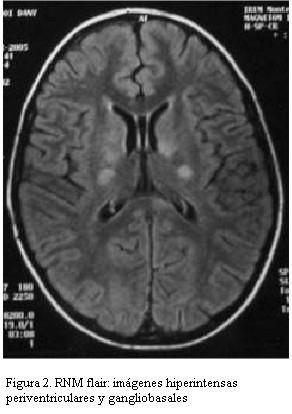
Se realiza tratamiento con ceftriaxona por 21 días y aciclovir por 72 horas hasta resultado de estudios virológicos, con regresión parcial de la sintomatología neurológica.
A los 3 meses reingresa con fiebre, aftas orales, dolor abdominal persistente, hematemesis, lesiones cutáneas vesículo–pustulosas en zona de punción y signos focales neurológicos caracterizado por: III par incompleto a derecha, VI y VII periférico a izquierda, signos de liberación piramidal en los cuatro miembros a predominio derecho y ataxia cerebelosa. Valoración oftalmológica normal. Estudio citoquímico de LCR: incoloro, límpido, proteinorraquia 1,19 g/dl, glucorraquia 0,38 g/dl, 18 GB (70% mononucleares). Cultivos estériles, virológico negativo.
RNM: extensas lesiones hiperintensas en T2 y Flair gangliobasal externo e interno derecho incluyendo cápsula interna con ligero efecto de masa. Lesión de tálamo a izquierda y lóbulos temporales a nivel de los uncus del hipocampo bilateral y cerebelo. Lesiones a nivel de ambos pedúnculos cerebrales, protuberancia y sector anterior de bulbo. Angiorresonancia: gruesos troncos arteriales de territorio cerebral anterior, media y posterior permeables, menor visualización de ramas arteriales secundarias que aparecen adelgazadas (figuras 3 y 4).
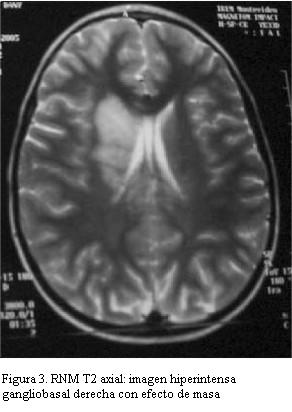
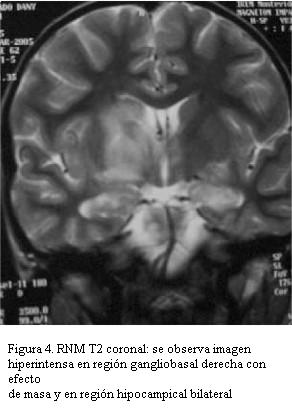
Con diagnóstico de enfermedad de Behçet se realizó corticoterapia, inicialmente metilprednisolona 30 mg/kg/día por 5 días y luego prednisona a 2 mg/kg/día, agregando a los 2 meses inmunosupresores (azatioprina) con descenso gradual de la corticoterapia. Se observó regresión completa de la lesión de pares craneanos, con persistencia de signos piramidales en miembros inferiores. No reiteró episodios de la esfera digestiva. Los potenciales evocados visuales, auditivos y de tronco fueron normales. Se realiza control oftamológico periódico, no presentando hasta el momento dolor ocular ni signos de uveítis.
La resonancia de control realizada a los 6 meses de iniciado el tratamiento mostró una franca mejoría de las lesiones.
Actualmente con 7 años, peso de 27 kg (P: 90–95), talla de 1,12 cm (P: 5–10), con síndrome cushingoide. Recibe en este momento azatiophrina 20 mg día (0,74 mg/kg/día) y prednisona a 25 mg días alternos. Con la disminución de la prednisona presenta episodios de fiebre y aftas orales que desaparecen al aumentar la dosis de corticoides a 25 mg extra, en el momento actual recibe dosis mínimas de corticoides en días alternos, desintometría ósea, fondo de ojo normales.
Discusión
La dispar prevalencia geográfica de la enfermedad y su asociación con el antígeno de histocompatibilidad HLA-B51 (B5) sugiere una predisposición genética.
La revisión más extensa de presentación en la edad pediátrica, sobre 86 casos constató una mayor frecuencia de casos familiares (15%), comparado con adultos (2%); este hecho es de particular interés para determinar el rol de factores genéticos y ambientales en la expresión de la enfermedad.
Algunos trabajos describen una frecuencia de 1/20.000 – 1/60.000 en edad pediátrica, sin clara diferencia entre ambos sexos (10-12). La lesión histopatológica corresponde a una vasculitis sistémica caracterizada por infiltración perivascular linfomonocitaria con o sin depósitos de fibrina en los vasos y necrosis tisular. La infiltración neutrofílica también se puede observar especialmente en las lesiones cutáneas. Estos hallazgos predominan en el sector venoso.
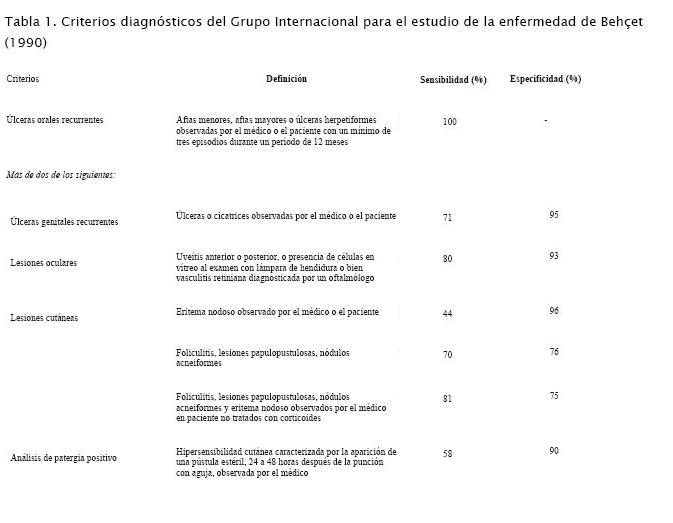
No existe un test específico para su diagnóstico, basándose sólo en criterios clínicos; los cuales fueron definidos en 1990 por el Grupo Internacional de estudio para la enfermedad de Behçet (tabla 1) (13). Este grupo internacional comenzó a trabajar en 1985 con 914 pacientes y seleccionó, como condición sine qua non, la presencia de aftas orales y las manifestaciones que unidas a las aftas orales permitieran diagnosticar la enfermedad; el uso de criterios mayores y menores radica en la prevalencia de síntomas y no en su gravedad ni especificidad. Si bien no hay ninguna lesión patognomónica, hay manifestaciones altamente sugestivas en pacientes que no reúnen todos los criterios: la patergia (hipersensiblilidad cutánea caracterizada por la aparición de pústula estéril 24 – 48 horas después de la punción cutánea con aguja), lesiones genitales cicatrizales, panuveítis, encefalitis de tronco y trombosis venosas (14).
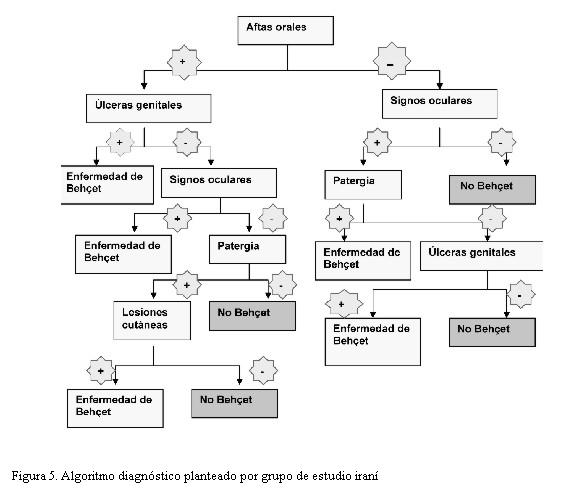
En nuestro caso, que debuta con aftas orales recurrentes y signos de encefalitis de tronco cerebral, si bien se podía sospechar esta enfermedad no cumplía en ese momento con los criterios del Grupo Internacional. En la revisión de Kané-Paut se constata que las lesiones neurológicas características son más frecuentes en los niños que no cumplen todos los criterios diagnósticos del grupo internacional. Estos criterios se cumplen en la evolución cuando agrega las lesiones cutáneas, signo de la patergia y el compromiso digestivo, esto está de acuerdo con la literatura. Destacamos que nuestro paciente no presentó en ningún momento úlceras genitales ni uveítis, los cuales son menos frecuentes en la edad pediátrica y que usualmente aparecen después de la pubertad. Por esta razón el grupo iraní de Barnes diseñó un algoritmo que permite diagnosticar al paciente con sólo dos manifestaciones, estableciendo un plan de tratamiento y vigilancia para prevenir la aparición de manifestaciones más graves de la enfermedad (figura 5) (14,15).
El compromiso del sistema nervioso central (SNC) que se observa entre 5–25% de los pacientes suele manifestarse en los cinco primeros años del debut de la enfermedad (16), como añadió nuestro caso. El compromiso de SNC se observa más frecuentemente en varones, dato coincidente con trabajos publicados. En un 5% de los casos puede ser la primera manifestación de la enfermedad. Si bien existen casos descritos de inicio en la primera infancia, en la serie pediátrica más extensa citada de 86 casos, la edad promedio de inicio fue de 8,4 años con un promedio de edad al realizar el diagnóstico de 13 años. La expresión neurológica se presenta en general con uno de los siguientes patrones: 1) meningoencefalitis crónica; 2) compromiso de tronco cerebral; 3) infarto cerebral; 4) hipertensión intracraneana secundaria a trombosis venosa cerebral. Kidd, Steuer y Denman en 50 casos con manifestaciones neurológicas encontraron un predominio de meningoencefalitis con compromiso de tronco cerebral (50%), como en nuestro caso (17). El compromiso neurológico puede ser de tipo parenquimatoso (neuro-Behçet) que se observa en el 80% de los casos y es de mal pronóstico, produciendo primariamente afectación del parénquima cerebral con tendencia a producir lesiones focales, especialmente en tronco cerebral, gangliobasal, cápsula interna y estructuras diencefálicas, o diseminada. La afectación no parenquimatosa que se observa en el 18% de los casos recibe el nombre de angio-Behçet o vásculo-Behçet, siendo en general de mejor pronóstico (18).
La resonancia magnética nuclear es el examen de elección para confirmar estas alteraciones, revelando en el neuro-Behçet tres patrones característicos: una fase inicial en la que se evidencian focos de hiperintensidad en T2 a nivel de tronco cerebral, gangliobasal y hemisferios cerebrales; una fase intermedia con mayor inflamación que causaría edema y efecto de masa que puede regresar espontáneamente o con tratamiento inmunosupresor, pudiendo encontrar focos de microhemorragia con hiposeñal por los depósitos de hemosiderina, y finalmente en la fase crónica predominan imágenes de atrofia especialmente en tronco encefálico. El seguimiento neuroimagenológico evidencia regresión o desaparición de las lesiones parenquimatosas a largo plazo (meses a años) (19).
El diagnóstico diferencial en esta forma de neuro-Behçet diseminada se realiza con procesos desmielinizantes típicos de la infancia como son la encéfalo mielitis difusa aguda, neuroborreliosis, esclerosis múltiple, neuro sweet, conectivopatías y otras vasculitis del SNC (20-22).
Debemos dejar claro que esta enfermedad es rara en nuestro país, sobre todo la forma neurológica. Pensamos que la consulta con el especialista en dermatología de nuestro paciente debería haber sido realizado tempranamente en la evolución.
El tratamiento de la enfermedad de Behçet es multidisciplinario, con médicos entrenados en el uso de inmunosupresores, el tratamiento recomendado para el paciente con compromiso neurológico son altas dosis de corticoides (metilprednisolona y prednisona) en la etapa aguda y posteriormente inmunosupresores (ciclosporina, azathioprine, ciclofosfamida, cloranbucil, talidomida). En nuestro caso estamos realizando tratamiento con azathioprine, dado que existen estudios controlados de la eficacia de este fármaco en esta enfermedad. Para la forma de angio-Behçet a este tratamiento se le añade la terapia anticoagulante y antiagregante (23-28).
En cuanto al pronóstico la enfermedad de Behçet se caracteriza por presentar un curso en empujes y remisiones. En la década de 1960 el neuro-Behçet tenía una alta mortalidad del 25% dentro del primer año de iniciadas las manifestaciones neurológicas. Actualmente otros trabajos muestran una mortalidad del 10–20% a los 5 años. Son factores de mal pronóstico: su inicio en la infancia, las alteraciones del líquido cefalorraquídeo y el compromiso del tronco cerebral
Las principales causas de muerte son: la hemoptisis masiva por arteritis pulmonar y trombosis venosa múltiple. La ceguera y enfermedad neurológica son las mayores causas de discapacidad permanente en esta enfermedad (2).
Referencias bibliográficas
1. Behçet H. Uber Rezidivierende, aphtose, dürch ein virus verursachte Geschwure am Munde, am Auge und an den Genitalien. Dermatologische. Wochenschrift 1937; 36: 1152–7.
2. Serdaroglu P. Behçet´s disease and the nervous system. J Neurol 1998; 245: 197–205.
3. Marshall S. Behçet´s disease. Best Pract Res Clin Rheumatol 2004; 18(3): 291-311.
4. Pereira M, Aleman J. Neuro Behçet. Bol Soc Peruana Med Interna 1996; 9 (3): 112-6.
5. López de Maturana D, Amaro P, Balestrini, Segovia L. Manifestaciones clínicas en 5 casos de enfermedad de Behçet. Rev Med Chile 2002; 130 (5): 551-6.
6. Sakane T, Takedo M, Suzuki N, Inaba G. Behçet’s Disease. N Engl J Med 1999; 341(17): 1285–91.
7. Calzada D, García J, Mustelier R, Fernandez A, Hernandez E. Neuro-Behçet. Presentación de tres casos. Rev Neurol 1999; 28(12): 1166–9.
8. Akman–Demir G, Serdaroglu P, Tasçi B; Neuro–Behçet Study Group. Clinical patterns of neurological involvement in Behçet disease: evaluation of 200 patients. Brain 1999; 122: 2171–81.
9. Magga D, Alayón C, Méndez E, De Anda G, Belloso A. Manifestaciones neurológicas de la Enfermedad de Behçet. Arch Med Interna 2003; 25 (2-3): 86-8.
10. Cheng Y, Thong B, Cheng H. Behçet´s Disease: Experience in Tertiary Rheumatology Center in Singapore and a Review of The literature. Ann Acad Med Singapore 2004; 33: 510-4.
11. Kone-Paut I, Yurdakul S, Bahabri S, Shafae N, Ozen S, Ozdogan H, et al. Clinical Features of Behçet´s disease in children: An international collaborative Study: 86 cases. J Pediatr 1998; 132: 721–5.
12. Uziel Y, Brik R, Padeh S, Barash J, Mukamel M, Harel L, et al. Juvenile Behçet´s disease in Israel. Clin Exp Rheumatol 1998; 16: 502–5.
13. International Study Group for Behçet´s Disease. Criteria for diagnosis of Behçet´s disease. Lancet 1990; 335: 1078–80.
14. Graña Gil J, Sánchez Meizozo M. Criterios diagnósticos y diagnóstico diferencial de la enfermedad de Behçet. Rev Clin Esp 2002; 202(1): 20–3.
15. Barnes CG. Behçet syndrome classification criteria. Ann Med Interne 1999; 150: 477-82.
16. Weiner S, Otte A, Schumacher M, Juengling F, Brink I, Nitzsche E, et al. Neuro-Behçet´s Syndrome in a Patient not Fulfilling Criteria for Behçet´s Disease: Clinical Features and Value of Brain Imaging. Clin Rheumatol 2000; 19: 231–4.
17. Kidd D, Steuer A, Denman A, Rudge P. Neurological complications in Behçet´s syndrome. Brain 1999; 122: 2183–94.
18. Akman Demir G, Bahar S, Coban O, Tasci B, Serdaroglu P. Cranial MRI in Behçet´s disease: 134 examinations of 98 patients. Neuroradiology 2003; 45: 851–9.
19. Gerber S, Biondi A, Dormont D, Wechsler B, Marsault C. Long-terms MR follow–up of cerebral lesions in neuro–Behçet´s disease. Neuroradiology 1996; 38: 761-8.
20. Al-Araji A, Sharquie K, Al-Rawi Z. Prevalence and patterns of neurological involvement in Behçet´s disease: a prospective study from Iraq. J Neurol Neurosurg Psychiatry 2003; 74: 608–13.
21. Coban O, Bahar S, Akman–Demir G, Tasci B, Yurdakul S, Yazici H, et al. Marked assessment of MRI finding: is it possible to differentiate Neuro – Behçet´s disease from other central nervous system. Neuroradiology 1999; 41: 255–60.
22. Hisanaga K, IwasaKi Y, Itoyama Y; The Neuro-Sweet Disease Study Group. Neuro-Sweet disease. Neurology 2005; 64: 1756–61.
23. Martínez A, Vidaller A, Mitjavila F, Pujol R, Jato M, Rubio F. Afectación parenquimatosa del sistema nervioso central en la enfermedad de Behçet. Rev Neurol 1998; 17 (156): 223–5.
24. Sakane T, Takeno M. Current Therapy in Behçet´s Disease. Skin Therapy Lett 2000; 5: 3-6.
25. Palacin V, Olalla R, Jimenez S, Rigau R, Civil F, Gonzalez C. Behçet´s disease therapy review. An Med Interna 2002; 19 (11): 594–8.
26. Bang D. Treatment of Behçet´s Disease. Yonsei Med J 1997; 38 (6): 401-10.
27. Kural–Seyahi E, Fresko I, Seyahi N, Ozyazgan Y, Mat C, Hamuryudan V, et al. The Long-Term Mortality and Morbility of Behçet´s Syndrome: A 2 - Decade Outcame Survey of 387 Patients Followed at a Dedicated Center. Medicine 2003; 82: 60–76.
28. Brik R, Shamali H, Bergman R. Successful Thalidomide Treatment of Severe Infantile Behçet Disease. Pediatr Dermatol 2001; 18: 143–5.
Correspondencia: Dr. Gabriel González.
Correo electrónico: visiongon@hotmail.com


{kind=link}
{kind=link}