










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.78 no.1 Montevideo Mar. 2007
CASO CLÍNICO
Arch Pediatr Urug 2007; 78(1)
Maldición de Ondina:
presentación de un caso clínico
Dres. Fernando Mañé Garzón 1, Víctor Raggio 2
1. Ex Profesor de Clínica Pediátrica. Profesor Emérito de la Facultad de Medicina.2. Prof. Adj. Departamento de Genética, Facultad de Medicina.
Fecha recibido: 13 de diciembre de 2006.
Fecha aprobado 13 de marzo de 2007.
Resumen
El síndrome de hipoventilación central congénita idiopático conocido como “maldición de Ondina” es una enfermedad poco frecuente caracterizada por un control anormal de la ventilación en ausencia de enfermedad pulmonar, neuromuscular, neurológica central o cardíaca evidenciable. Múltiples genes se han demostrado asociados a esta afección y a otros fenotipos relacionados en su patogenia, como la enfermedad de Hirschsprung. Se presenta un caso demostrativo de esta afección y de las dificultades de manejo que ocasiona. Se discuten aspectos de su genética, diagnósticos diferenciales y mecanismos patogénicos.
Palabras clave:
APNEA DEL SUEÑO CENTRAL
HIPOVENTILACIÓN
CRESTA NEURAL
ENFERMEDAD DE HIRSCHSPRUNG
SÍNDROMES DE LA APNEA DEL SUEÑO
DIAGNÓSTICO DIFERENCIAL
GENÉTICA
Summary
Central, congenital, idiopathic hypoventilation syndrome known as ¨Ondine´s Curse¨, is a non frequent disease characterized by an abnormal ventilation control in the absence of pulmonary, neuromuscular, neurological or cardiac disease. Multiple genes are associated with this disease as well as to other related phenotypes such as Hirschsprung disease. A case showing its treatment difficulties is presented. Genetic aspects, etiology and differential diagnosis are exposed.
Key words:
SLEEP APNEA, CENTRAL
HYPOVENTILATION
NEURAL CREST
HIRSCHSPRUNG DISEASE
SLEEP APNEA SYNDROMES
DIAGNOSIS, DIFFERENTIAL
GENETICS
Introducción
El sindrome de hipoventilación central congénita idiopático (SHCCI), también conocido como “maldición de Ondina” (OMIM 209880) es una enfermedad poco frecuente caracterizada por un control anormal de la ventilación en ausencia de enfermedad pulmonar, neuromuscular, neurológica central o cardíaca evidenciable (1). La primera descripción fue realizada por Mellins y colaboradores en 1970 (2). Se trata de un defecto primario del control autonómico que resulta en una inadecuada (o ausente) respuesta ventilatoria a la hipoxia y/o la hipercapnia, presente desde las primeras horas de vida (3). En general los pacientes tienen una adecuada ventilación despiertos, pero hipoventilan durante el sueño de onda lenta; en algunos casos más severos la hipoventilación se da tanto durante el sueño como la vigilia (4).
La denominada “maldición de Ondina” ha sido tomada de la clásica obra de Friedrich La Motte Fouqué, Ondine, publicada en 1811 (5), en la cual la protagonista, un hada llamada Ondina, hija de las aguas, al contraer amor con Hans, un mortal, si éste la engaña él muere y ella pierde la memoria de su amante. Él muere y al ser maldecido, le condena a morir por no respirar sino en vigilia. Esto ocurre luego de una trama de fascinantes situaciones. Esta leyenda del folclore germano inspiró a Jean Giraudoux una obra teatral publicada en 1939 (6), que ha ingresado al repertorio teatral, tanto francés como universal.
El SHCCI se asocia a un grupo heterogéneo de alteraciones conocidas genéricamente como “neurocrestopatías” (7), es decir fenotipos anormales surgidos por un defecto primario en la migración de células de la cresta neural. Entre éstas se incluyen: neuroblastoma, ganglioneuroma y la enfermedad de Hirschsprung (EH), o megacolon agangliónico, que aparece hasta en un 20% de los pacientes con SHCCI (8).
En relación a la genética, la investigación en SHCCI ha ido de la mano de la de la EH y del concepto referido de “neurocrestopatías” (9). Si bien la mayoría de los casos son esporádicos, se han descrito varios casos de aparición familiar vertical por lo que se plantean como modelos genéticos más probables formas con genes de efecto mayor así como herencia oligogénica, en la que mutaciones en pocos genes combinadas pueden producir el fenotipo patológico (10,11).
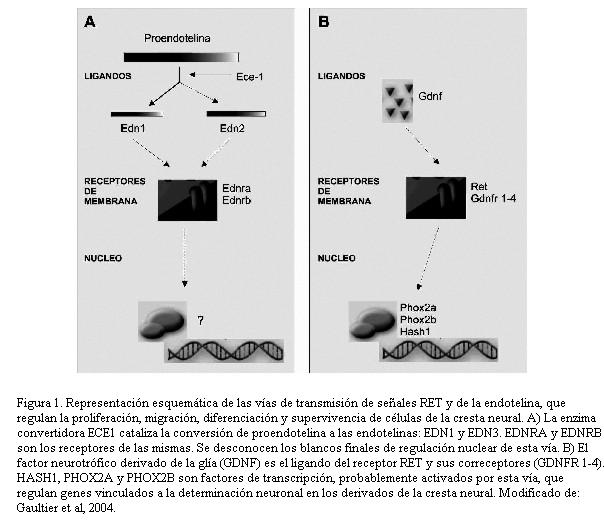
La búsqueda de genes causantes de estas afecciones ha sido más fructífera cuando se consideraron fenotipos amplios y procesos patológicos más que entidades clínicas estrictas: la hipótesis central de esta búsqueda es que el SHCCI es la forma más severa de defectos autonómicos primarios, que se pueden expresar en una gama de fenotipos (12), y que esta afección podía explicarse por alteraciones en las vías de transducción de señales que regulan el desarrollo de la cresta neural: la vía de RET y de la endotelina (figura 1). De este modo se han encontrado varios genes asociados a ellos (13).
En la EH se ha determinado que mutaciones en el gen RET dan cuenta del 50% de los casos de EH familiar y del 20% de los casos de EH esporádica (14,15). Otros de los genes que se han demostrado involucrados son el de la endotelina (EDN3) y el de su receptor (EDNRB) (16) que dan cuenta de aproximadamente 5% de los casos de EH. Se ha demostrado que mutaciones en la endotelina 3 (17) y RET (18) pueden manifestarse como SHCCI. Mutaciones en el gen del factor neurotrófico derivado del cerebro (BDNF) pueden causar tanto EH como SHCCI así como, en casos con una expresividad clínica más leve, hipotensión ortostática y síncope vasovagal (19). El gene PHOX2B es necesario para el desarrollo del sistema nervioso autónomo (SNA) y mutaciones en el mismo causan también SHCCI con un modo de herencia autosómico dominante (20), así como otros defectos autonómicos y tumores del SNA (neuroblastoma) (21). Una de las mutaciones descritas en este gene es una expansión de polialaninas (22), que, como en muchas otras patologías, cuanto mayor es la expansión más severa es la expresión clínica de la patología (23). Otro de los genes codificadores de factores de transcripción asociados a esta afección es HASH1 (24).
Por lo tanto, se ha determinado la participación de genes vinculados a tres vías de señalamiento intercelular; además, en todos los casos hay una penetrancia incompleta en los heterocigotos para estas mutaciones y una muy amplia variabilidad en la expresión clínica. Todo esto apoya el modelo de una herencia oligogénica interactiva en el SHCCI. En otros casos, mutaciones en un único gene pueden causar la afección, con una herencia dominante y penetrancia incompleta (25,26). Además, no se puede descartar que un evento secundario (ambiental desconocido, genes modificadores o mutaciones adicionales de novo), sean necesarios para la expresión completa de este síndrome. PHOX2B parece ser el principal gen etiológico en esta afección, tanto en términos de frecuencia como de severidad clínica, y la amplísima variabilidad clínica estaría explicada por la concurrencia de otras mutaciones y, eventualmente, otros factores modificadores (ref. 11).
Por su singular interés presentamos un caso de SHCCI demostrativo de la misma y de las dificultades de manejo que ocasiona. Asimismo se enfatiza la variable expresión clínica de este tipo de defectos que debe hacer pensar en un espectro de disfunciones autonómicas primarias que el clínico debe tener en cuenta.
Caso clínico
Recién nacido de sexo femenino, producto de segunda gestación, padres no consanguíneos, sin antecedentes familiares a destacar. Embarazo bien controlado, sin complicaciones. Parto vaginal eutócico, de término, adecuado para la edad gestacional: peso al nacer: 3350 g, longitud: 50 cm, perímetro cefálico: 36 cm, Apgar 5/7, depresión neonatal moderada. Se realizó ventilación con ambú, recuperando frecuencia cardíaca a los 3 minutos. Pobre esfuerzo respiratorio, se colocó bajo Hood. A los primeros minutos de vida sufre apneas por lo que se realiza intubación orotraqueal. Se extuba a las dos horas. Durante la internación reitera episodios de cianosis y apneas que se recuperan espontáneamente. Reitera apnea grave que requiere estímulo y ventilación con ambú, quedando hipotónica, pálida e hiporreactiva. Episodio de movimientos tónico-clónicos que se interpretan como convulsión. Se le realiza fenobarbital y se ingresa a CTI.
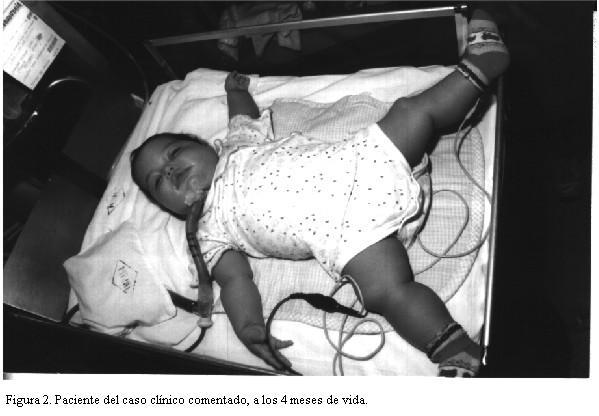
Al examen (figura 2) se presentaba pálida, leve cianosis de labios y extremidades, mal perfundida, hiporreactiva, hipotonía global, temperatura axilar 36ºC, pupilas midriáticas, reflejo fotomotor presente, fontanela anterior de 2 por 1 cm, normotensa. Sin malformaciones externas.
Abdomen distendido, depresible, sin visceromegalias. Cardiovascular: ritmo regular de 120 cpm, sin soplos, pulsos presentes. Pleuropulmonar: intubada, regular entrada de aire bilateral, estertores crepitantes basales bilaterales.
Exámenes complementarios: glicemia: 0,94 g/l; gasometría arterial: pH 7,4, pCO2 16, pO2 190, BE -11, HCO3 10,7; Radiografía de tórax: morfológicamente normal. PCR: negativa. Hemocultivo: negativo. Electroencefalograma (EEG): sin signos patológicos.
En la evolución persiste intubada con bajos parámetros respiratorios, en coma (Glasgow 3), muy hipotónica. En reiterados intentos de extubación, pasándose previamente a CPAP, se obtienen muy escasos movimientos ventilatorios espontáneos con gasometrías que muestran franca hipoxemia e hipercapnia severa (de hasta 164 mmHg) por lo que no se puede retirar de AVM. Relevos infeccioso y genético-metabólico normales. Resonancia nuclear magnética (RNM) de cráneo muestra signos de inmadurez cerebral propios de la edad, sin patología encefálica focal. Radiografía de columna cervical: normal. Se realizó alimentación parenteral hasta el noveno día de vida, cuando se comienza alimentación progresiva por gastroclisis. Al mes y veinte días de vida se diagnostica estenosis hipertrófica del píloro que se resuelve quirúrgicamente por pilorotomía extramucosa. Múltiples episodios de infecciones respiratorias bacterianas. A los tres meses EEG que evidencia: sufrimiento cerebral generalizado moderado, sin signos focales. Múltiples episodios convulsivos que se interpretan como desencadenados por hipoxia, constatándose actividad epileptógena en EEG. Intentos de ventilación espontánea durante aproximadamente una hora al día, que se van prolongando progresivamente, pero persistiendo en AVM. A los 8 meses de edad se realiza nueva RNM que muestra imagen compatible con “arborización” de la sustancia blanca a nivel paracentral y occipital, no apreciándose aún signos de mielinización de la sustancia blanca a nivel parietal como es habitual a esta edad. Sin lesiones parenquimatosas focales. Mínimo aumento de los espacios subaracnoideos de la convexidad y del sistema ventricular y de cisternas. No se evidencian alteraciones vasculares. Persiste con episodios de apnea breves, de recuperación espontánea y crisis convulsivas a pesar del tratamiento con múltiples anticomiciales. Reitera episodios de neumonías agudas intrahospitalarias y un trastorno funcional de la deglución (por lo que hasta ese momento había sido alimentada por sonda orogástrica). Se realiza gastrostomía. Al año y 7 meses: crecimiento adecuado, retardo de las conductas motoras con aceptable desarrollo en el área social y coordinación, imposible valorar lenguaje. Potenciales visuales: permeabilidad funcional de la vía visual para la luz difusa, conservada bilateralmente, sin signos de sufrimiento.
Dependiente de la ventilación, alimentación por gastrostomía. Recibiendo por su epilepsia: carbamacepina, valproato y vigabatrin.
Broncodisplasia y varias neumonías (cultivándose secuencialmente: Staphylococcus aureus y gérmenes Gram negativos, estafilococo coagulasa negativo, Proteus mirabilis y Pseudomona aeruginosa). Superados estos múltiples episodios se intentan desconexiones temporales del ventilador de entre 15 minutos y una hora. Al año y 8 meses: nuevo episodio de neumonía por Staphylococcus aureus, hipercapnia y caídas de la saturación de O2; tratamiento con vancomicina y meropenem. En radiografía de tórax: opacidad homogénea en vidrio esmerilado de ambos campos pulmonares. Recibe inotrópicos. Agravación clínica que lleva al paro cardiorrespiratorio; se constata fallecimiento tras reanimación inefectiva.
En suma, se trata de un paciente de sexo femenino, quien presenta una severa hipoventilación desde el nacimiento, que es permanente pero empeora notoriamente durante el sueño (llevando a apneas que no revierten espontáneamente). Las complicaciones de la hipoxia episódica y de la intubación orotraqueal a permanencia, con múltiples episodios de neumonía a gérmenes multirresistentes, marcaron su evolución negativa que llevó a la muerte.
Comentarios
Las características clínicas de este caso nos llevan a formular el diagnóstico de síndrome de Ondine, por presentar un síndrome de hipoventilación central congénita idiopática, con falla severa del control central de la ventilación desde el nacimiento sin otra causa demostrable que lo explique. Los criterios diagnósticos comúnmente utilizados (27) para este diagnóstico clínico, que la paciente presenta, son: 1) evidencia persistente de hipoventilación durante el sueño (PCO2 > 60 mmHg); 2) inicio de los síntomas antes del año de vida; 3) ausencia de enfermedad pulmonar o neuromuscular primarias que puedan explicar la hipoventilación; 4) ausencia de enfermedad cardíaca primaria.
Como diagnósticos diferenciales se deben descartar en primer lugar las causas adquiridas de síndrome de hipoventilación, fundamentalmente las de origen en lesiones encefálicas perinatales hipóxico-isquémicas. En este caso no hubo tales antecedentes y la RNM de cráneo no mostró lesiones focales. Además, el deterioro de la ventilación fue inmediato al nacimiento y estable. De todos modos no se puede descartar una influencia de la medicación antiepiléptica y una posible lesión hipóxica encefálica postnatal (de la que contamos con un EEG patológico, que informa: “sufrimiento cerebral generalizado moderado”).
En relación a la fisiopatología, la falta de sensibilidad central a la hipercapnia e hipoxia parece responder a una falla de los mecanismos neuronales que integran los datos de los quimiorreceptores con los centros del control de la respiración. Algunos pacientes tienen alteraciones autonómicas más amplias como: alteraciones del ritmo cardíaco, dismotilidad esofágica, sudoración excesiva, HD o tumores de la cresta neural. En esta paciente se constató una alteración de la deglución por lo que se debió alimentar por gastrostomía.
Según los datos actuales la gran mayoría de los casos de SHCCI están causados por mutaciones en PHOX2B, y en menor medida en otros genes causales. En el caso de esta paciente no se extrajo una muestra de ADN previo a su fallecimiento por lo que no contamos con la posibilidad de realizar un diagnóstico molecular a nivel de los genes mencionados como causantes de esta afección.
El espectro fenotípico de las mutaciones en estos genes, especialmente RET es amplísimo (28), por lo que hay que estar atento a otros fenotipos en la familia como EH, alteraciones autonómicas, alteraciones respiratorias de expresión más tardía, que puedan indicar a portadores de mutaciones. Por otro lado, las variantes en estos genes podrían subyacer a la reportada variabilidad interindividual en las apneas de los recién nacidos así como al síndrome de muerte súbita infantil. Es posible que el conocimiento de estos aspectos referidos a la genética del control de la respiración, guíen nuevas aproximaciones terapéuticas y preventivas a estos problemas (29).
Referencias bibliográficas
1. Costa Orsay JA, Pons M. Síndrome de Ondine: diagnóstico y seguimiento. Na Pediatr (Barc) 2005; 63(5): 426-32.
2. Mellins RB, Balfour HH, Turino GM, Winters RW. Failure of automatic control of ventilation (Ondine’s curse). Medicine (Baltimore) 1970; 49: 487–504.
3. Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gautier C, et al. The French Congenital Central Hypoventilation Syndrome Registry. General Data, Phenotype, and Genotype. Chest 2005; 127: 72–9.
4. Weese-Mayer DE, Shannon DC, Keens TG, Silvestri JM. American Thoracic Society statement on the diagnosis and management of idiopathic congenital central hypoventilation syndrome. Am J Res Crit Care Med 1999; 160: 368–73.
5. La Motte Fouqué F. Ondine. Berlin: Hatiers, 1811.
6. Giraudoux J. Ondine, piece en 3 actes. Paris: Grassets, 1939.
7. Bolande RP. The neurocristopathies: a unifying concept of disease arising in neural crest maldevelopment. Hum Path 1974; 5: 409-29.
8. Croaker GD, Shi E, Simpson E, Cartmill T, Cass DT. Congenital central hypoventilation syndrome and Hirschsprung’s disease. Arch Dis Child 1998; 78(4): 316-22.
9. Godbole K. The Many Faces of Hirschsprung’s Disease. Indian Pediatrics 2004; 41: 1115-23.
10. Marazita ML, Maher BS, Cooper ME, Silvestri JM, Huffman AD, Smok-Pearsall SM, et al. Genetic segregation analysis of autonomic nervous system dysfunction in families of probands with idiopathic congenital central hypoventilation syndrome. Am J Med Genet 2001; 100(3): 229-36.
11. Weese-Mayer DE, Silvestri JM, Marazita ML, Hoo JJ. Congenital central hypoventilation syndrome: inheritance and relation to sudden infant death syndrome. Am J Med Gene 1993; 47: 360-7.
12. Weese -Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet 2003; 123(3):267-78.
13. Gaultier C, Amiel J, Dauger S, Trang H, Lyonnet S, Gallego J, et al. Genetics and Early Disturbances of Breathing Control. Pediatr Res 2004; 55: 729–33.
14. Attie T, Pelet A, Edery P, Eng C, Mulligan L, Amiel J, et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 1995; 4: 1381–6.
15. Sakai T, Wakizaka A, Matsuda H, Nirasawa Y, Itoh Y. Point Mutation in Exon 12 of the Receptor Tyrosine Kinase Proto-oncogene RET in Ondine-Hirschsprung Syndrome. Pediatrics 1998; 101: 924-5.
16. Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell 1994; 79(7): 1257-66.
17. Bolk S, Angrist M, Xie J, Yanagisawa M, Silvestri JM, Weese-Mayer DE, et al. Endothelin-3 frameshift mutation in congenital central hypoventilation syndrome. Nat Genet 1996; 13: 395–6.
18. Sakai T, Wakizaka A, Matsuda H, Nirasawa Y, Itoh Y. Point mutation in exon 12 of the receptor tyrosine kinase proto-oncogene RET in Ondine-Hirschsprung syndrome. Pediatrics 1998; 101(5): 924-6.
19. Weese-Mayer DE, Bolk S, Silvestri JM, Chakravarti A. Idiopathic congenital central hypoventilation syndrome: evaluation of brain-derived neurotrophic factor genomic DNA sequence variation. Am J Med Genet 2002; 107: 306-10.
20. Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 2003; 33: 1–3.
21. Trochet D, O’Brien LM, Gozal D, Trang H, Nordenskjold A, Laudier B, et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet 2005; 76(3):421-6.
22. Amiel J, Laudier B, Attie´-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-liked homeoboxgene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 2003; 33: 459–61.
23. Matera I, Bachetti T, Puppo F, Di Duca M, Morandi F, Casiraghi GM, et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset central hypoventilation syndrome. (Letter). J Med Genet 2004; 41: 373-80.
24. De Pontual L, Nepote V, Attie-Bitach T, Al Halabiah H, Trang H, Elghoussi V, et al. Noradrenergic neuronal development is impaired by mutation of the proneural HASH-1 gene in congenital central hypoventilation syndrome (Ondine’s curse). Hum Mol Genet 2003; 12: 3173–80.
25. Bolk S, Angrist M, Schwartz S, Silvestri JM, Weese-Mayer DE, Chakravarti A. Congenital central hypoventilation syndrome: mutation analysis of the receptor tyrosine kinase RET. Am J Med Genet 1996; 63: 603-9.
26. Amiel J, Salomon R, Attie T, Pelet A, Trang H, Mokhtari M, et al. Mutations of the RET-GDNF signaling pathway in Ondine’s curse. (Letter). Am J Hum Genet 1998; 62: 715-7.
27. Gozal D. Congenital Central Hypoventilation Syndrome: an update. Pediatr Neumol 1998; 26: 273-82.
28. Marazita ML, Berry-Kravis EM, Silvestri JM, Weese Mayer DE. Congenital Central Hypoventilation Syndrome [on line] GeneReviews set. 2006 <www.geneclinics.org> [consulta: set.2006].
29. Gozal D. New concepts in abnormalities of respiratory control in children. Curr Opin Pediatr 2004; 16(3): 305-8.
Correspondencia: Prof. Dr. Fernando Mañé Garzón. Departamento de Historia de la Medicina, Facultad de Medicina. Gral. Flores 2125. CP 11800. Montevideo, Uruguay
Correo electrónico: histmed@fmed.edu.uy


{kind=link}
{kind=link}