










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.77 no.2 Montevideo June 2006
CASO CLÍNICO
Arch Pediatr Urug 2006; 77(2): 160-167
Raquitismo vitamina D dependiente tipo I
Dras. Margarita Halty 1, Marina Caggiani 2
1. Ex Asistente de Clínica Pediátrica. Intensivista de Niños. Nefrólogo. Pediatra Nefrólogo de la Policlínica Nefrológica del CHPR.2. Ex Prof Adj de Clínica Pediátrica C. Pediatra Nefrólogo de la Policlínica Nefrológica del CHPR. Docente Honorario de la Facultad de Medicina UDELAR. Clínica Pediátrica C Prof. Dra. I. Rubio. Centro Hospitalario Pereira Rossell. Facultad de Medicina. UDELAR.
Fecha recibido: 26 de junio 2006.
Fecha aprobado: 30 de junio 2006.
Resumen
La enfermedad ósea que se expresa como raquitismo deriva de diferentes desórdenes, desde la carencia de vitamina D, síndromes malabsortivos, enfermedad renal crónica, displasia metafisaria, hipofosfatasia y raquitismos resistentes, dentro de los cuales están los hipofosfatémicos y aquellos vitamina D dependientes.
Presentamos una paciente de 2 años y 6 meses, con síntomas y signos de un raquitismo activo, hipocalcemia severa asintomática, normofosforemia, aumento de la fosfatasa alcalina y de la hormona paratiroidea y con niveles normales de 25 hidroxi vitamina D en sangre. Se diagnosticó como un probable raquitismo vitamina D dependiente tipo I dada la respuesta satisfactoria y rápida con calcio y 0,5 microgramos/día de calcitriol por vía oral.
Palabras clave: RAQUITISMO
CALCITRIOL-deficiencia
CALCITRIOL-uso terapéutico
Summary
Rickets is a disease which disturbs normal bone formation through different methods, like vitamin D deficiency, malabsorption, chronic renal disease, metaphisary dysplasia, low phosphorus and resistant rickets.
A two and a half year old patient with active rickets who had asymptomatic severe hypocalcemia, normal phosphorus level, high alkaline phosphatase and high parathyroid hormone and normal serum 25- hydroxycholecalciferol. Probable vitamin D dependence type I rickets was diagnosed due to her rapid recovery with the treatment given.
Key words: RICKETS
CALCITRIOL-deficiency
CALCITRIOL-therapeutic use
Introducción
El raquitismo es la afección ósea resultante de la deficiente mineralización del hueso o del tejido osteoide en crecimiento. En esta situación, existe un retardo del crecimiento y de la calcificación normal del cartílago epifisario. Los osteoblastos secretan colágeno para formar la matriz que luego no se mineraliza. Concomitantemente se produce la reabsorción de hueso y de la matriz por los osteoclastos (1).
La vitamina D3 (7- dehidrocolesterol) presente en la piel se activa a colecalciferol por vía fotoquímica y se hidroxila en el hígado a 25 hidroxicolecalciferol (calcidiol o 25 OH D) y luego en la corteza renal a 1,25 dihidrocolecalciferol (calcitriol o 1,25 (OH)2 D). Este último es el metabolito activo, cuyas acciones son:
- en el intestino, facilitar la absorción de calcio y fósforo;
- en el riñón, estimular la reabsorción de ambos;
- en el hueso, producir el depósito mineral y el turnover del mismo (1).
La calcificación del osteoide depende de la presencia de niveles adecuados de calcio y de fósforo en el líquido extracelular. Dichos niveles están influidos por la vitamina D cuya acción máxima depende de su activación a través de las hidroxilaciones mencionadas.
La clínica y la radiología en los diferentes tipos de raquitismo pueden ser similares, aunque con diferencias en la edad de presentación y en la severidad de la enfermedad. Los exámenes de laboratorio serán la piedra angular para diferenciar los distintos trastornos.
El raquitismo carencial se presenta a edad temprana y con una intensidad variable de acuerdo al grado de déficit vitamínico. Se caracteriza a nivel humoral por un valor normal o levemente disminuido del calcio sérico, una hipofosfatemia y un incremento de la fosfatasa alcalina (FA). El nivel de 25 hidroxicolecalciferol está descendido y el de la hormona paratifoidea (PTH) aumentado. Otros hallazgos comprenden una alteración en la capacidad de acidificación renal, pudiendo encontrarse también hiperfosfaturia, glucosuria y aminoaciduria.
Los síndromes malabsortivos generan carencia de vitamina D y un perfil humoral similar al carencial. Deben orientarse siempre los estudios a buscar este tipo de enfermedad.
Una vez descartados el raquitismo carencial y los trastornos malabsortivos, otras osteopatías incluyen diferentes desórdenes que pasamos a analizar (1-5):
1) Raquitismo hipofosfatémico familiar. Consiste en un defecto a nivel renal para reabsorber suficiente fosfato. Se evidencia a partir de los 6-10 meses de edad, en que los niveles de fósforo descienden por debajo de 3,5 mg%. Cursa con calcemia normal y sin hiperparatiroidismo. Se trata de un defecto en el transporte tubular proximal de fosfato y de la 1 alfa hidroxilación renal de la vitamina D. Se transmite en general como un rasgo dominante ligado al X, pero también hay casos autosómicos recesivos o dominantes.
2) Síndrome de Fanconi. Se produce una expoliación de fosfato a nivel renal, junto con aminoaciduria y glucosuria, siendo las causas genéticas o adquiridas.
3) Raquitismo vitamina D dependiente tipo I. Resulta de una deficiencia genética en la enzima que convierte el calcidiol (25 OH D) en calcitriol (1,25 (OH)2 D) en el riñón por un déficit de la enzima 1 alfa hidroxilasa. La herencia es autosómica recesiva y el gen se sitúa en el cromosoma 12. Las alteraciones clínicas y de laboratorio son más severas que las del raquitismo carencial. Los pacientes presentan una hipocalcemia grave, siendo variable la concentración de fosfato (normal o levemente descendido), elevación de la fosfatasa alcalina y de la PTH. La dosificación de 1,25 (OH)2 D sérica muestra valores muy descendidos o aun inexistentes, los niveles de 25 OH D son normales o elevados.
4) Raquitismo vitamina D dependiente tipo II o defecto del receptor o raquitismo vitamina D resistente. Se debe a un defecto heredado en forma recesiva a nivel del receptor del calcitriol. La mitad de los pacientes presentan alopecia. A nivel humoral cursan con alteraciones similares a las del grupo anterior pero con niveles de 1,25(OH)2 D muy elevados en sangre.
5) Defecto de la 25 hidroxilasa. Se han reportado sólo dos casos hasta el momento, la herencia es probablemente recesiva.
6) Osteodistrofia. En la insuficiencia renal extrema ocurre un déficit de 1 alfa hidroxilasa y disminución en la excreción renal de fosfato. Cursa con hipocalcemia y es el único raquitismo con un nivel de fosfato elevado.
7) Hipofosfatasia. Se caracteriza por la ausencia de fosfatasa alcalina. Genera raquitismo sin disturbios en el metabolismo del calcio y del fósforo.
8) Displasia metafisaria. Condición genética de carácter dominante con una alteración a nivel del cromosoma 6. Las características radiológicas son similares a las del raquitismo carencial, pero con mayor severidad a nivel de la metáfisis proximal del fémur. No cursa con alteraciones en los valores de calcio, fósforo, fosfatasa alcalina ni de los metabolitos de la vitamina D en sangre (1-5).
Excepto la hipofosfatasia y la displasia metafisaria, todos los disturbios tienen en común el presentar una elevación de la fosfatasa alcalina (1).
Objetivo
Comunicar un caso de raquitismo severo en una niña de 2 años de etiología muy poco frecuente y por tal motivo con dificultades diagnósticas, que probablemente corresponda a un raquitismo vitamina D dependiente tipo I, con una respuesta inicialmente favorable al tratamiento.
Caso clínico
Preescolar de 2 años y 6 meses, sexo femenino, raza blanca, procedente de Las Piedras, de un MSE aceptable. Madre 40 años, sana (talla 1,57 m), padre 47 años (talla 1,73 m), sano, no consanguíneos. Hermana de 4 años, sana.
Antecedentes personales: producto de tercer embarazo (un aborto espontáneo), mal tolerado por diabetes gestacional, recibió tratamiento dietético. Cesárea a las 40 semanas por cesárea previa, PN 3.670 g, PC 35 cm, T 47 cm, Apgar 9/10.
Alimentación con pecho directo hasta los 2 años, sólidos desde los 6 meses, actualmente completa. Recibió vitamina D durante 2 meses, exposición al sol en forma esporádica, últimamente fue mayor por indicación médica. Buen progreso ponderal: al año peso 10.160 g (percentil 75), talla 72,5 cm (percentil 25). Al año y 10 meses, peso 12.000 g (entre percentil 50 y 75) y talla 75.5 cm (por debajo del percentil 3). A los 2 años y 3 meses peso 12.000 g (entre percentil 25 y 50) y talla 78 cm (por debajo del percentil 3). A los 2 años y 6 meses, peso 12.250 g (por encima del percentil 25) y talla 79 cm (por debajo del percentil 3). Velocidad de crecimiento de los últimos 3 meses: 4 cm/año.
Marcha al año y 4 meses, con dificultad y caídas frecuentes. Buena motricidad fina. Lenguaje adecuado.
A partir del año de edad, la madre realizó varias consultas por dificultad al incorporarse desde la posición en decúbito, trastorno de la marcha con caídas frecuentes, deformación torácica e incurvación de miembros inferiores. Notó además fatigabilidad fácil al deambular y sudoración profusa, fundamentalmente nocturna.
No presentó nunca síntomas digestivos. Deposiciones normales. No anorexia.
A los 2 años de edad, se inició el estudio por hipocrecimiento y marcha patológica. Dadas las deformaciones óseas, se derivó al Servicio de Ortopedia donde se realizaron radiografías y desde donde la derivaron a la Policlínica de Nefrología del Centro Hospitalario Pereira Rossell.
Consultó en la Policlínica de Nefrología a los 2 años y 3 meses, destacándose del examen: buen estado general, buen vínculo con la madre y el entorno de acuerdo a su edad, juega. Piel sana, mucosas bien coloreadas. A nivel osteoarticular: frente olímpica, cabeza en forma de caput quadratum, fontanela anterior permeable, de 3 x 2 cm, normotensa; rosario costal, cincha de Harrison; pulseras radiales; rodetes supramaleolares. Deformaciones de miembros inferiores, con arqueamiento de los mismos y coxa vara (figura 1). Hipotonía muscular, abdomen prominente en la estación de pie, lordosis lumbar. Inestabilidad en la marcha. La dentición era adecuada para la edad.
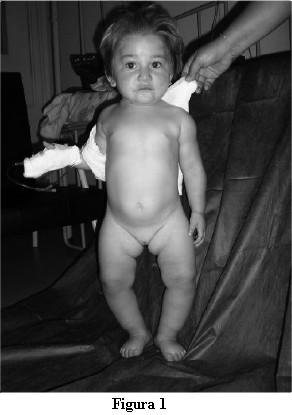
El resto del examen no presentaba nada a destacar.
Con diagnóstico clínico de raquitismo severo, se solicitó la evaluación correspondiente que mostró una hipocalcemia severa, normofosforemia, elevación de la FA y aumento de la PTH. Se inició tratamiento ambulatorio con calcio por vía oral, que se aumentó en forma progresiva sin respuesta (hipocalcemia persisente). Del resto de los exámenes se destacaban: una acidosis metabólica leve, examen de orina y función renal normales, hemograma normal y funcional hepático normal. Se solicitó la dosificación sérica de los metabolitos de la vitamina D, sólo se realiza la de 25 OH D3 en nuestro medio, siendo el resultado normal. La excreción urinaria de calcio era normal y la de fósforo se encontraba aumentada (índice fósforo/creatinina = 2,9 siendo el valor normal menor de 2).
Calcio total: 6,2 mg/dl. Iónico: 0,72 mEq/l
P: 4,3 mg/dl.
FA: 6015 UI/l (VN para la edad: < 673).
PTH: 717 pg/ml (VN: 14-72).
25 OH D3 32,6 ng/ml (VN 9–37,6).
Gasometría pH 7,34 HCO3- 18,9 mEq/l BE –6,7.
Na: 141 mEq/l.
K: 3,8 mEq/l.
Cl: 103 mEq/l.
Orina : normal.
PEF y metabolismo del hierro normales.
Anticuerpos antitransglutaminasa y antiendomisio negativos.
IgA sérica 13,2 mg/dl (VN: 26- 150).
Ecografía renal normal.
Las radiografías óseas mostraron una descalcificación global, los huesos largos incurvados y ensanchamiento de las metáfisis de los mismos en forma de copa (figuras 2 y 3).
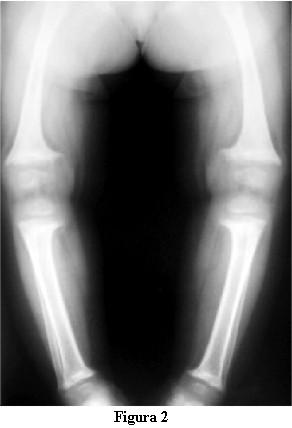
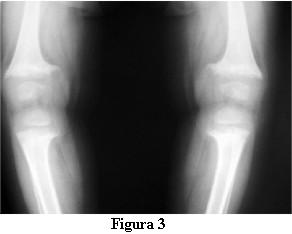
Se decidió su ingreso al hospital en el momento de iniciar el tratamiento con calcitriol v/o, para control estrecho del medio interno.
Se inició la administración de calcio intravenoso, bajo la forma de gluconato, a una dosis de 1.000 mg/m2 de superficie corporal por día, hasta estabilizar la calcemia. A las 48 horas, con una calcemia de 8,2 mg/dl se indicó el calcitriol v/o. Al 4º día se descendió lentamente la dosis de calcio i/v hasta su suspensión mientras se iniciaba e incrementaba la administración del mismo por vía enteral.
Se administró 1,25 (OH)2 D3 por v/o, inicialmente a una dosis de 0,125 mg/día, que se incrementó hasta 0,5 mg/día.
En la evolución en sala nunca tuvo signos de hipocalcemia; la sudoración disminuyó a la semana del tratamiento. La fontanela anterior se redujo 1 cm en sus diámetros a los 15 días, y al mes estaba casi cerrada. Luego de retirada la infusión continua de calcio, que la obligaba a permanecer en reposo, pudo reiniciar la deambulación, que fue progresiva, fundamentalmente luego del alta, sin acusar fatigabilidad.
A nivel humoral, mejoraron los parámetros bioquímicos. La calcemia se mantuvo siempre dentro de los valores bajos de la normalidad.
Presentó hipofosforemia transitoria que mejoró espontáneamente. La fosfaturia se normalizó luego de iniciado el tratamiento con vitamina D.
Presentó hipomagnesemia que se corrigió por vía intravenosa.
Tuvo acidosis metabólica leve con compensación respiratoria que retrocedió parcialmente sin tratamiento. El anión gap en sangre fue 16 y el urinario fue positivo, mostrando un déficit en la excreción de amonio. Nunca recibió bicarbonato vía oral por la dificultad en su administración, por tal motivo no pudo estudiarse la bicarbonaturia.
La internación duró 16 días, al alta recibía 0,5 mg de calcitriol en una dosis y 6 g de calcio distribuidos en el día. La dosis de calcio se descendió en forma ambulatoria, de acuerdo a los controles de la calcemia hasta 2 g al mes de iniciado el tratamiento, y actualmente, a los 3 meses, recibe 1 g al día.
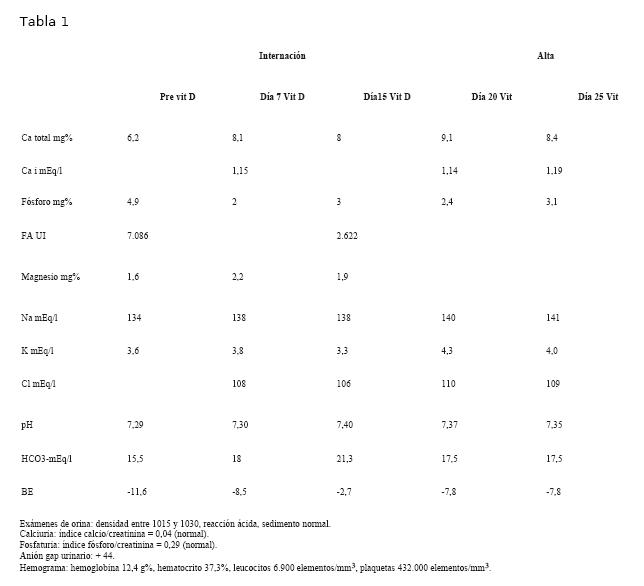
La tabla 1 muestra una síntesis de los valores humorales durante la internación y al alta.
En la evolución a los 3 meses de iniciado el tratamiento con calcitriol, a los 2 años y 9 meses, la marcha tiene las dificultades derivadas de las deformaciones de los miembros pero no presenta caídas, ni fatigabilidad, ni sudoración. La talla es de 81 cm , por debajo del percentil 3. La velocidad del crecimiento de los últimos 3 meses es de 8 cm/año. La fontanela anterior está completamente cerrada.Las radiografías óseas (figuras 4 y 5) muestran, a pesar del corto tiempo de evolución, una disminución del signo de la copa, con un progreso de la diáfisis hacia el tejido osteoide.
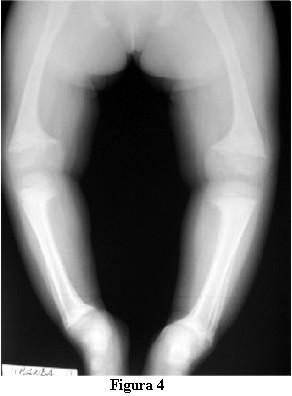
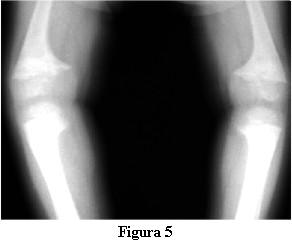
Del resto de los exámenes actuales se destacan: calcio iónico: 1,20 mEq/l; FA 1443 UI/l.
Discusión
Vimos el caso de una preescolar de 2 años, con hipocrecimiento y deformidad progresiva de miembros inferiores que motivaron la consulta. De la anamnesis surgen, además, síntomas como fatigabilidad al deambular escasos metros y sudoración profusa. Lo anterior, sumado al aspecto general, con las deformaciones descritas a nivel de cráneo, miembros inferiores, metáfisis de huesos de miembros y costillas, nos conduce al diagnóstico de raquitismo severo, que las radiografías confirmaron.
Si bien la ingesta de vitamina D puede haber sido insuficiente durante el primer año de vida, nos encontramos ante una niña de 2 años, con elementos de raquitismo activo en todas las etapas evolutivas. Esto nos orientó hacia un tipo de raquitismo diferente al carencial, motivo por el cual no iniciamos de inmediato el tratamiento del mismo sin catalogarlo.
El perfil humoral con una hipocalcemia severa desbordaba también la posibilidad de un raquitismo carencial, así como la concentración sérica normal de fósforo y la dosificación normal de 25 OH D.
Era improbable que se tratara de un síndrome malabsortivo, en ausencia de síntomas digestivos, sin otras carencias nutricionales, calórico–proteicas, ni de minerales. Los anticuerpos antiendomisio y antitransglutaminasa fueron negativos en presencia de una concentración de IgA sérica descendida, lo que dificultaba la interpretación. La clínica, la ausencia de otras carencias nutricionales y la buena respuesta a la 1,25 (OH)2 D nos alejaron este diagnóstico.
Nos orientamos hacia otro tipo de raquitismo, vitamina D resistente, que se refiere a aquel que no responde al tratamiento habitual para el raquitismo carencial con vitamina D2 (6).
Dentro de los raquitismos resistentes, un primer grupo está constituido por los hipofosfatémicos, y el segundo por los seudodeficientes o vitamina D dependientes tipo I y II (6). En este último grupo podemos incluir el defecto de la 25 hidroxilasa (2).
El raquitismo hipofosfatémico familiar se caracteriza por hipofosforemia, normocalcemia y dosificación normal de PTH. El hiperparatiroidismo puede desarrollarse en esta entidad secundariamente al tratamiento con fósforo (3-5).
La fosforemia normal, la hipocalcemia severa y el hiperparatiroidismo en nuestra paciente descartaban la posibilidad de la hipofosfatemia familiar ligada al cromosoma X.
El síndrome de Fanconi también se acompaña de hipofosforemia debido a las pérdidas tubulares de fósforo, acompañado de glucosuria y aminoaciduria. En sangre presentan hipofosfatemia y normocalcemia (1).
El perfil humoral y urinario de la niña alejaban la posibilidad de este diagnóstico, aunque inicialmente la fosfaturia fue elevada, pero se normalizó con el tratamiento. No se comprobaron otras pérdidas tubulares, la acidosis metabólica fue de grado leve, secundaria al raquitismo. La fosfaturia inicialmente alta podría deberse a la acción de la PTH; posteriormente la fosfaturia descendió. Esto puede explicarse por una inhibición secundaria en la secreción de PTH debido al aumento de la calcemia, por el descenso del fósforo sérico por depósito óseo y por la acción del calcitriol.
En el segundo grupo de raquitismos vitamina D resistentes la presentación clínica es la de un raquitismo con alteraciones radiológicas severas de los huesos largos, en presencia de hipocalcemia grave, nomofosfatemia, hiperparatiroidismo secundario y gran aumento de la fosfatasa alcalina. Este perfil humoral puede hallarse en cualquiera de las 3 formas mencionadas: defecto de la 25 hidroxilasa, raquitismo vitamina D dependiente tipo I (RVDD I) y raquitismo vitamina D dependiente tipo II (RVDD II).
El defecto de la 25 hidroxilasa, la forma más rara de las tres, se descartó en la paciente por la dosificación de 25 OH D sérica, que estaba en un valor normal alto.
El diagnóstico diferencial entre las otras dos formas (RVDD I y RVDD II) se hubiera aclarado en forma rápida y definitiva mediante la dosificación de 1,25(OH)2 D, recurso del que carecemos en nuestro medio.
Entonces el diagnóstico definitivo fue empírico, tras evaluar la respuesta al tratamiento.
Ambas entidades son infrecuentes, siendo los datos bibliográficos escasos para el RVDD I y más numerosos para el RVDD II.
El RVDD I, además de todas las características antes citadas, puede presentar hipoplasia del esmalte dental, la dosificación de 25(OH) D es normal o elevada y la de 1,25 (OH)2 D está en un nivel muy bajo o indetectable, debido a un defecto en la 1 alfa hidroxilación renal. La respuesta es rápida y completa con el aporte exógeno de la vitamina activa o de 1 OH D.
La causa del mismo es una mutación en el gen responsable de la 1 alfa hidroxilación que fue estudiada por S.J. Smith y colaboradores. Resulta de un defecto en el componente del citocromo P450c1. La proteína en juego (CYP27B1) es codificada por un gen en el cromosoma 12q13.1-13.3.
Otros órganos como la placenta, los queratinocitos, las sinoviales, el peritoneo, los pulmones y los macrófagos sanguíneos también presentan actividad de 1alfa hidroxilasa, pero su acción es local, para la función inmune y para mantener la homeostasis del calcio a nivel tisular. La actividad de la enzima a nivel renal estaría codificada por el mismo gen que la de la placenta y los queratinocitos, en cambio la enzima de los demás tejidos dependería de uno diferente (7).
J. Silver y colaboradores compararon dos pacientes con RVDD. Uno de ellos tenía importantes trastornos e incapacidad en la marcha, los niveles de 1,25 (OH)2 D eran bajos y fue interpretado como tipo I. Se trató en forma satisfactoria con 1microgramo diario de 1OH D. El otro paciente presentaba alopecia, tenía niveles de 1,25 (OH)2 D elevadísimos y no se obtuvo ninguna respuesta con igual dosis de vitamina. En éste se hizo diagnóstico de RVDD tipo II (8).
K. Widhalm y colaboradores también se plantearon el diagnóstico diferencial entre los tipos I y II de RVDD en un paciente con raquitismo severo. Dado que no contaban con dosificación sérica de metabolitos de vitamina D, le administraron un tratamiento con dosis bajas de 1OH D. El mismo fue exitoso, lo que los indujo a hacer diagnóstico de tipo I (9).
En el trabajo de E.E. Delvin y colaboradores, se estudiaron nueve pacientes con raquitismo vitamina D dependiente tipo I, ocho de los cuales cursaban con hipocalcemia y siete con hipofosforemia leve, todos ellos con elevación de los niveles de PTH. Se comprobó la existencia de niveles bajos de 1,25 (OH)2 D. Se empleó una dosis de calcitriol entre 0,25 y 2 mg/día lográndose el retorno a los valores humorales normales a los 4 meses. A los que se había practicado biopsia ósea, la misma se reiteró luego de los 9 meses, mostrando mejoría en la mineralización ósea. Los autores sugieren un monitoreo estrecho de las variables bioquímicas, dado que los requerimientos de calcitriol varían durante el curso del tratamiento (10).
Otros autores manejan dosis similares para el tratamiento, entre 0,5 y 1,5 mg/ día por v/o (2).
El RVDD II es una enfermedad hereditaria, autosómica recesiva que consiste en una alteración en el sistema receptor- efector a la 1,25 (OH)2 D en los órganos blanco. A diferencia de la entidad anterior, ésta puede presentarse con alopecia en la mitad de los casos y a nivel humoral, los niveles de 1,25 (OH)2 D en sangre son elevadísimos. El resto de la presentación clínica y humoral es en todo similar. La etiopatogenia explica además otra diferencia: la respuesta al tratamiento con vitamina activa es incierta y ocurre con dosis muy elevadas; las necesarias para elevar la calcemia y hacer retroceder el raquitismo oscilan entre 3–6 y hasta 12,5 mg/ día de 1 OH D o de 1,25 (OH)2 D. Si hay alopecia, ésta no mejora con el tratamiento. Debe controlarse la calciuria durante el tratamiento prolongado con metabolitos de la vitamina D, ya que la misma puede elevarse por hiperabsorción intestinal de calcio y/o por supresión de la PTH (11-13).
Pueden existir diferencias en el grado de resistencia al tratamiento con calcitriol o con 1 OH D, probablemente debido a diferentes defectos del receptor. Algunos efectos de la vitamina dependen de acciones rápidas o no genómicas, y otras lentas o genómicas, pero todas son mediadas por el receptor de la vitamina D (RVD) (12,14).
Se han descrito diferentes tipos de mutaciones que alteran la estructura y la función del RVD. La mutación 1268T determina un cambio del aminoácido isoleucina por treonina, alterando el dominio de ligand- binding, zona del RVD que interactúa directamente con la 1,25 (OH)2 D. El RVD 1268T mutante tiene 5 a 10 veces menor afinidad por el calcitriol (15).
Otra mutación en Helix 12 afecta el sitio de unión a los coactivadores, se trastorna entonces la interacción con éstos (16). Se halló también una única inserción/sustitución en el dominio de unión del receptor, por mutación en Helix H1 (17).
La sustitución del aminoácido triptofano por arginina (W286R) en el RVD determina la abolición de la respuesta del gen blanco a la vitamina, no produciéndose la inducción normal del RNA m para la 24- hidroxilasa (18).
Otra mutación del RVD se denomina H305Q y causa una disminución de la afinidad por el receptor, por lo que el tratamiento requiere dosis elevadísimas de calcitriol (13).
En todos los casos el tratamiento consiste en mantener dosis suprafarmacológicas de 1,25 (OH)2 D durante períodos prolongados, requiriendo incluso a menudo administración parenteral de la misma y calcio oral o parenteral.
El grado de respuesta del raquitismo al tratamiento probablemente depende del defecto exacto en la función del RVD (19).
En la paciente estudiada se descartaron otros tipos de trastornos del metabolismo óseo:
La función renal (filtrado glomerular) y la ecografía renal eran normales, por lo que no se trataba de una osteodistrofia renal.
Los disturbios humorales descartaron el diagnóstico de hipofosfatasia y de displasia metafisaria (1).
A la luz de la bibliografía se planteó entonces que la paciente era portadora de un raquitismo dependiente de la vitamina D, normofosfatémico y con una dosificación normal –alta de 25 OH D. Las posibilidades diagnósticas se redujeron entonces a 2: RVDD I o RVDD II. Dado que no contábamos con la dosificación de 1,25 (OH)2 D en sangre, la respuesta a la administración exógena de la misma nos daría el dato empírico del tipo de raquitismo. La bibliografía menciona que son suficientes dosis de 0,25 a 2 mg/día para un tratamiento satisfactorio del RVDD I, en contraste con las dosis mucho más elevadas que se requieren en el tipo II. La dosis baja de calcitriol empleada de 0,5 mg/día y la rápida respuesta al tratamiento nos permitieron inferir una buena sensibilidad del RVD y que se trataba entonces de un RVDD I.
La certeza diagnóstica definitiva la hubiera aportado la dosificación de 1,25 (OH)2 D en sangre, si se encontraba en niveles bajos o indetectables. De todas formas, el proceso de curación clínico, radiológico y humoral nos aseguró que el tratamiento era el correcto. Se controlará en la evolución la necesidad de una intervención por parte de un ortopedista para la alineación de sus miembros inferiores. De no mejorar espontáneamente la acidosis se la puede corregir mediante la administración de un álcali.
El tratamiento debe ser seguido con controles clínicos y humorales, para adaptarlo a los requerimientos variables de calcio y calcitriol en una paciente en crecimiento.
Nuestro objetivo es lograr la curación y devolverle así a la niña su potencial óptimo de crecimiento y desarrollo que estaban afectados con su enfermedad no tratada.
Referencias bibliográficas
1. Heird WC. Vitamin Deficiencias and Excesses. En: Behrman RE, Kliegman RM, Jenson HB. Textbook of Pediatrics. 17 ed. Philadelphia: Saunders, 2004: 186-9.
2. Finberg L. Metabolic Bone Disease [en línea] Medicine dec 18 2003 <http.//www.emedicine.com> [consulta: 10 ene 2006].
3. Chaussain-Miller C, Sinding C, Wolikow M, Lasfargues JJ, Godeau G, Garabédian M. Dental abnormalities in patients with familial hypophosphatemic vitamin D- resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr 2003; 142: 324-31.
4. Yamamoto T. Diagnosis of X-linked hypophosphatemic vitamin D resistant rickets. Acta Paediatr Jpn 1990; 39: 499-502.
5. Ono T, Seino Y. Medical management and complications of X-linked hypophosphatemic vitamin D resistant rickets. Acta Paediatr Jpn 1997; 39: 503-7.
6. Garabedian M. Vitamin D resistant rickets [en línea] Orphanet jan 2002 <http://www.orpha.net> [consulta: 10 ene 2006].
7. Smith SJ, Rucka AK, Berry JL, Davies M, Mylchreest S, Paterson CR, et al. Novel Mutations in the 1 alpha – Hydroxylase (P450c1) Gene in Three Families with Pseudovitamin D- Deficiency Rickets Resulting in Loss of Functional Enzyme Activity in Blood- Derived Macrophages. J Bone Miner Res 1999; 14(5): 730-9.
8. Silver J, Landau H, Bab I, Shvil Y, Friedlaender MM, Rubinger D, et al. Vitamin D- dependent rickets types I and II. Diagnosis and response to therapy. Isr J Med Sci 1985; 21(1): 53-6.
9. Widhalm K, Waldhauser F, Zwiauer K. Transient vitamin- D dependent rickets? a case report differential diagnosis. Padiatr Padol 1982; 17(2): 409-16.
10. Delvin EE, Glorieux FH, Marie PJ, Pettifor JM. Vitamin D dependency: replacement therapy with calcitriol? J Pediatr 1981; 99(1): 26-34.
11. Takeda E, Yokota I, Saijo T, Kawakami I, Ito M, Kuroda Y. Effect of Long-Term Treatment with Massive doses of 1 alpha –Hydroxyvitamin D3 on Calcium- Phosphate Balance in Patients with Vitamin D- Dependent Rickets Type II. Acta Paediatr Jpn 1990; 32: 39-43.
12. Kruse K, Feldmann E. Healing of rickets during vitamin D therapy despite defective vitamin D receptors in two siblings with vitamin D –dependent rickets type II. J Pediatr 1995; 126: 145-8.
13. Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary Vitamin D Resistant Rickets Caused by a Novel Mutation in the Vitamin D Receptor That Results in Decreased Affinity for Hormone and Cellular Hyporesponsiveness. J Clin Invest 1997; 99: 297-304.
14. Nguyen TM, Lieberherr M, Fritsch J, Guillozo H, Alvarez ML, Fitouri Z, et al. The Rapid Effects of 1,25- Dihydroxyvitamin D3 Require the Vitamin D Receptor and Influence 24- Hydroxylase Activity. J Biol Chem 2004; 279: 7591- 7.
15. Malloy PJ, Xu R, Peng L, Peleg S, Al–Ashwal A, Feldman D. Hereditary 1,25-Dihydroxyvitamin D Resistant Rickets due to a Mutation Causing Multiple Defects in Vitamin D Receptor function. Endocrinology 2004; 145: 5106- 14.
16. Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A Novel Mutation in Helix 12 of the Vitamin D Receptor Impairs Coactivator Interaction and Causes Hereditary 1,25- Dihydroxyvitamin D-Resistant Rickets without Alopecia. Mol Endocrinol 2002; 16: 2538-46.
17. Malloy PJ, Xu R, Cattani A, Reyes ML, Feldman D. A Unique Insertion/Substitution in Helix H1 of the Vitamin D Receptor Ligand Binding Domain in a Patient With Hereditary 1,25-Dihydroxyvitamin D-Resistant Rickets. J Bone Miner Res 2004; 19: 1018-24.
18. Nguyen TM, Adiceam P, Kottler ML, Guillozo H, Rizk-Rabin M, Brouillard F, et al. Tryptophan Missense Mutation in the Ligand- Binding Domain of the Vitamin D Receptor Causes Severe Resistance to 1,25 Dihydroxyvitamin D. J Bone Miner Res 2002; 17: 1728-37.
19. Langman CB. Disorders of Phosphorus, Calcium and Vitamin D. En: Barratt TM, Avner ED, Harmon WE. Pediatric Nephrology. 4 ed. Baltimore : Lippincott Williams & Wilkins, 1999: 529-44.
Correspondencia: Dra. Margarita Halty
E-mail: margahalty@hotmail.com


{kind=link}