










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.77 no.2 Montevideo jun. 2006
CASO CLÍNICO
Arch Pediatr Urug 2006; 77(2): 154-159
Ataxia-teFlangiectasia (síndrome de Louis-Bar)
Dres. Marcos Delfino 1, Rina Bruzzone 2, Andrea Rey 3, Aurora Delfino 4, Ma. Catalina Pírez 5
1. Pediatra. Ex Residente de Pediatría.2. Postgrado de Neuropediatría.
3. Neuropediatra. Ex Asistente de Neuropediatría.
4. Ex Profesora de Neuropediatría.
5. Profesora Clínica Pediátrica del CHPR.
Clínica Pediátrica. Cátedra de Neuropediatría. Centro Hospitalario Pereira Rossell (CHPR).
Fecha recibido: 28 de abril de 2006
Fecha aprobado: 26 de junio de 2006
Resumen
Se presenta el caso clínico de una enfermedad pediátrica poco frecuente. Se trata de una niña de 7 años con un desarrollo neurológico alterado y sintomatología cerebelosa que agrega síntomas respiratorios crónicos a la que se le diagnostica síndrome de Louis-Bar (ataxia-telangiectasia). Se le realizó paraclínica tendiente a confirmar dicho diagnóstico y se inició el tratamiento paliativo disponible. Falleció a los 9 años de edad.
Se revisa la etiopatogenia y los criterios diagnósticos de la enfermedad.
Palabras clave: ATAXIA TELANGIECTASIA
ENFERMEDADES RESPIRATORIAS
PROGRESIóN DE ENFERMEDAD
Summary
The clinical case of an uncommon pediatric illness is presented. A 7 year old girl with an abnormal neurological development and cerebellum symptoms who added chronic respiratory symptoms was diagnosed with Louis-Bar syndrome (ataxia-telangiectasia).
Laboratory studies were performed in order to confirm diagnosis, and available palliative treatment was started. She died at the age of 9. Etiology and diagnostic criteria are reviewed.
Key words: ATAXIA TELANGIECTASIA
RESPIRATORY TRACT DISEASES
DISEASE PROGRESSION
Introducción
La ataxia-telangiectasia es un síndrome que fue descrito por primera vez en 1926 por Syllaba y Henner (1). Sin embargo, fue la publicación de la neuróloga Denise Louis-Bar en 1941 (2) la que tuvo mayor repercusión en el medio médico y generó la asociación del síndrome a su nombre. La denominación ataxia-telangiectasia llegó por fin con la detallada descripción por parte de Boder y Sedgwick en 1959 (3). Desde entonces otros autores, e incluso múltiples talleres internacionales, han investigado este síndrome, cuyo interés ha aumentado en la medida de conocerse su sustrato molecular. En Uruguay, existen las publicaciones de la doctora Rebollo en 1961(4) y del doctor Julio Marcos y colaboradores 1962 (5).
Se trata de una enfermedad poco frecuente (1 caso cada 40 a 100 mil nacimientos) de herencia autosómica recesiva que se transmite a través del cromosoma 11, siendo los portadores (heterocigotos) aparentemente sanos. Un 1 a 2% de la población es portadora (6,7).
Clínicamente se manifiesta por ataxia cerebelosa progresiva, distonía, telangiectasias oculocutáneas, retraso en el crecimiento, envejecimiento prematuro, infecciones sinopulmonares recidivantes, riesgo aumentado de cáncer (en particular linfoide) e hipersensibilidad a las radiaciones ionizantes. Otras manifestaciones clínicas pueden ser hipogonadismo y retraso del advenimiento de la pubertad, y diabetes sacarina no cetósica y resistente a la insulina. En 1988 Gatti y colaboradores (8) identificaron la región cromosómica 11q22-23 como la implicada en este síndrome.
En 1995 el grupo de Silo (9) identificó en esa región el gen mutado de la ataxia-telangiectasia (ATM). El producto de ese gen resultó ser una proteinquinasa con funciones de “señal” que interviene en el control del ciclo celular, la recombinación de ADN, la apoptosis y otras respuestas celulares al daño de ADN. Las anormalidades en este control mediado por ATM producen ADN dañado, con lo cual se acumulan las roturas cromosómicas con el paso del tiempo y la célula muere. Estos fenómenos afectarían especialmente los timocitos, los linfocitos B inmaduros, las células de Purkinje del sistema nervioso central y el endotelio vascular (10).
La marcha atáxica puede estar presente en el segundo año de vida; a los cuatro años de vida la presentará el 85% de los afectados. La ataxia afecta progresivamente la musculatura axial, de las extremidades, del paladar surgiendo así disartria, babeo, apraxia ocular, coreoatetosis. A los 10 años de vida la marcha independiente es imposible.
Las telangiectasias surgen posteriormente, entre los 4 y los 8 años de edad en escleróticas y piel (orejas, mejillas, pliegues de codos, etcétera). La hipersensibilidad cutánea a la luz se evidencia por un envejecimiento temprano de la piel (10,11). La inmunodeficiencia celular y humoral es responsable de la recurrencia de las infecciones respiratorias altas y bajas, estas últimas conducen a enfermedad pulmonar crónica. Las neumonías infecciosas (virales o bacterianas) o por broncoaspiración constituyen la primera causa de muerte en estos enfermos (10).
La segunda causa de muerte son los cánceres. El 38% de los pacientes con esta enfermedad desarrollan un neoplasma en su vida. Esto puede ser debido a las translocaciones cromosómicas y roturas excesivas del ADN que caracterizan la inestabilidad genética de esta enfermedad. Un 85% de estos neoplasmas son de origen hematológico. La mayoría son tumores linforreticulares, desarrollando uno de cada 100 afectos un nuevo tumor anual a partir de los 10 años. Generalmente se trata de linfomas (60%) y leucemias agudas (27%), aunque después de los 20 años la frecuencia de estos cánceres cae un 50%, siendo más frecuentes los tumores de origen epitelial. La hipersensibilidad a la radiación ionizante debida a la inestabilidad genética hace que los estudios imagenológicos en estos pacientes deban ser restringidos y que las dosis de radiación para tratar estos tumores sean menores a las usuales.
El riesgo de cáncer (especialmente de mama) entre portadores no está aún confirmado. Estudios realizados por Swift et al (12) sugieren que hasta un 5% de todos los pacientes que mueren de cáncer menores de 45 años podrían ser portadores del gen ATM (heterocigotos). Las madres de los pacientes con síndrome de Louis-Bar, portadoras del gen ATM, tendrían un riesgo incrementado para cáncer de mama (10,11).
El timo puede ser indetectable desde la primera infancia y si está presente es pequeño y carece de diferenciación córticomedular; lasz amígdalas son hipoplásicas, así como el tejido linfoide periférico. En la sangre hay linfopenia progresiva tanto para los linfocitos T como B; los linfocitos T CD4+ (timodependientes) mueren selectivamente, lo que finalmente conduce a una inversión de la normal relación CD4-CD8. Las funciones de uno y otro disminuyen gradualmente, lo que sería también un efecto de la excesiva rotura de ADN. Los defectos de tipo humoral incluyen deficiencia de inmunoglobulinas. Existe un déficit de IgA tanto a nivel sérico como de la IgA secretoria (por ejemplo, saliva) en el 60% de los casos. La IgG2 disminuye en el 80%; también pueden disminuir la IgE, las isohemaglutininas y las respuestas de anticuerpos específicos (10,11).
La edad de fallecimiento es generalmente alrededor de los 20 años; casi todos los pacientes mueren antes de los 39 años (10,11,13).
La dificultad que generó el diagnóstico debido a su rara ocurrencia y sus manifestaciones multisistémicas, así como las posibilidades terapéuticas limitadas hasta el momento a medidas paliativas, otorgan gran interés al presente caso clínico.
Caso clínico
M. M., 7 años, sexo femenino, procedente de Artigas, medio socioeconómico y jurídico deficitario. Padre desconocido, sin antecedentes familiares a destacar. Producto de primer embarazo y parto normales, recién nacido sin patología. Enlentecimiento de la adquisición de conductas motoras. La madre nota inestabilidad en la sedestación aproximadamente un año antes de la internación, en forma progresiva inestabilidad en la marcha, actualmente marcha inestable con aumento de la base de sustentación.
No concurre a la escuela. Certificado Esquema de Vacunación: vigente.
Desde noviembre de 2001 síntomas respiratorios: tos, expectoración y fatiga.
Múltiples episodios de aumento de la tos y fatiga acompañados de fiebre que se interpretan como neumonías. Concomitantemente repercusión nutricional, adelgazamiento de 4 kg en 4 meses.
Consulta el 26 de febrero de 2002 por exacerbación de su sintomatología respiratoria con fiebre. Examen físico: peso: 13,5 kg (P<3), talla: 1,07 m (P<3), perímetro craneano: 51 cm (P 50). Lúcida, mal aspecto general, fascies inexpresiva, disminución global del panículo adiposo, disminución de masas musculares, decaída, frecuencia respiratoria 28 rpm sin tiraje.
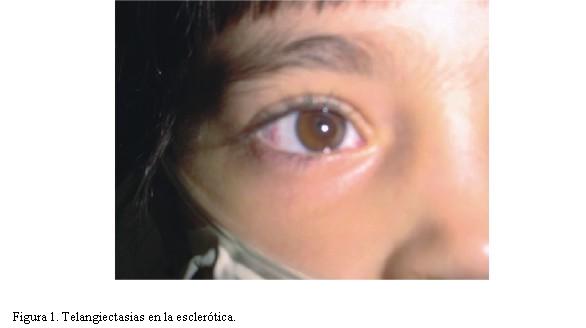
Hipocratismo digital. Piel y mucosas normocoloreadas, teleangiectasias a nivel conjuntival bilateral (figura 1). No se observan estas lesiones en la piel. Tórax simétrico sin remodelación, sonoridad conservada, estertores subcrepitantes difusos bilaterales.
Cardiovascular: ritmo regular de 136 cpm, tonos bien golpeados, no soplos. Presión arterial: 90/60 mm Hg.
Del examen psiconeuromuscular destacamos:
- Apraxia oculocefálica: imposibilidad de mirar hacia un objeto sin desplazar la cabeza.
- Pares craneanos: hipomimia facial sin otros elementos patológicos en el resto de pares craneanos. No disartria ni babeo.
- Sector espinal: fuerzas conservadas, extensibilidad y pasividad aumentadas en los cuatro miembros, reflejos osteotendinosos disminuidos, temblor fino distal.
- Coordinación: discronometría bilateral.
- Sensibilidad: conservada.
- Estática: aumento de la base de sustentación, retropulsión danza tendinosa del tibial anterior. Maniobra de Romberg negativa. Marcha inestable, con aumento de la base de sustentación.
Estudios complementarios
Saturación de oxígeno ventilando espontáneamente al aire: 91%.
Hemograma 26/2/02: glóbulos blancos (GB): 16.200 elementos/mm3 (segmentados 60%, linfocitos 20%); hemoglobina (Hb): 12,1 g/dl, hematocrito (Hto): 36,3; plaquetas: 690.000 elementos/mm3.
Velocidad de eritrosedimentación: 37 mm/h.
Radiografía de tórax: hiperinsuflación, con infiltrado intersticial difuso.
Tomografía Axial Computarizada de tórax: normal.
Alfafetoproteína (AFP): 319,7 ng/ml (valor de referencia: hasta 10 ng/mL).
Estudio oftalmológico: telangiectasias en escleróticas, resto normal (figura 1).
Proteinograma electroforético:
- Albúmina: 3,39 g/dl (normal: 3,14-4,70 g/dl).
- Alfa 1: 0,32 g/dl (normal: 0,14-0,38 g/dl).
- Alfa 2: 0,74 g/dl (normal: 0,59-1,01 g/dl).
- Beta: 0,70 g/dl (normal: 0,61-0,95 g/dl).
- Gamma: 0,35 g/dl (normal: 0,95-1,65 g/dl).
Dosificación de inmunoglobulinas (Ig):
- Ig A: <23,1 g/dl (normal: 30-240 g/dl).
- Ig E: <17,8 g/dl (normal: hasta 90 g/dl).
- Ig G: <146 g/dl (normal: 610-1.380 g/dl).
- Ig M: 427,1 g/dl (normal: 20-134 g/dl).
Subclases de Ig G:
Ig G1: <94 g/dl (normal: 400-1.080 g/dl).
Ig G2: 17 g/dl (normal: 85-410 g/dl).
Ig G3: 9 g/dl (normal: 15-149 g/dl).
Estudio inmunofenotípico en sangre: se analizó por citometría de flujo muestra de sangre anticoagulada con EDTA, utilizándose para el estudio inmunofenotípico los siguientes anticuerpos monoclonales: kit comercial lymphogramR (que incluye: CD4, CD3, CD56, CD8, CD19) e, independientemente, CD45, CD14, CD45RA y CD62L.
Resultados: las poblaciones linfocitarias analizadas se encontraron dentro de los valores de referencia para la edad (VR), salvo dos subpoblaciones: a) aquellas correspondientes a células natural killer (NK) CD56+ cuyo valor porcentual (22,6%) se encontró por encima del valor utilizado (VR mediana: 9,82%, desvío estándar: 5,01%)(14); b) más significativo aún fue el porcentaje hallado (0,17%) de linfocitos T cooperadores “naive” (CD4+/CD45RA+/CD62L+) el cual se encuentra muy por debajo de los valores esperados (VR_ mediana: 26%, percentiles 5-95: 17-40%), tanto para valores de referencia en adultos como en niños (15).
Tratamiento
Sintomático. M. M. requirió O2 entre 0,5 y 1,5 l/min. a permanencia. La saturación de O2 pasó a 93-94%, permaneciendo O2 dependiente hasta su muerte. Se realizó apoyo nutricional con complementos alimenticios, a pesar de lo cual no se logró mejorar su crecimiento. Las infecciones respiratorias sucesivas se interpretaron siempre como bacterianas y se trataron con cefalosporinas de tercera generación y/o ciprofloxacina. Se redujeron los estudios imagenológicos al mínimo imprescindible y las internaciones se abreviaron lo más posible. Recibió gammaglobulina intravenosa profiláctica a dosis de 400 mg/k una vez al mes, lo que no impidió que las infecciones respiratorias recidivaran con la misma frecuencia. El 20/4/04 fallece en su domicilio en Artigas a los 9 años de edad. No se realizó necropsia.
Discusión
El diagnóstico de ataxia-telangiectasia en esta niña de 7 años se fundamenta en el florido cuadro clínico y los resultados de laboratorio.
El grupo de Lederman del Ataxia-telangiectasia Clinical Center propone como criterios diagnósticos(16): I) hallazgos neurológicos característicos: a) marcha atáxica en los primeros 2 o 3 años de edad; b) signos oculomotores y disartria; c) trastornos asociados del movimiento, ataxia progresiva, hipomimia, trastornos de la deglución y neuropatía periférica. II) al menos uno de los siguientes: a) telangiectasia ocular; b) nivel elevado en sangre de alfa fetoproteína; c) ruptura cromosómica espontánea o inducida por rayos X.
El error diagnóstico más frecuente es la confusión con la parálisis cerebral(16). La única ataxia asociada a elevación de alfa fetoproteína es la ataxia-telangiectasia, hecho conocido hace más de treinta años(17).
Como puede observarse, la inmunodeficiencia no se incluye entre los criterios diagnósticos dada su ausencia en hasta un tercio de los casos.
Vista la importancia de un diagnóstico temprano (en particular para el consejo genético) y para evitar estudios radiológicos en el paciente afectado, el mismo debería basarse en la presencia de alfa fetoproteína elevada en un paciente con ataxia cerebelosa de inicio temprano, sin esperar la aparición de la telangiectasia a los 3 o 6 años (16).
Nuestra paciente presentaba: historia de enfermedad neurológica progresiva e infecciones respiratorias recurrentes; retraso en el crecimiento con importantes elementos de desnutrición; apraxia óculocefálica, síndrome cerebeloso (vermiano y hemisférico) y telangiectasias oculares. Presentó, por otra parte, una concentración de alfa fetoproteína sérica treinta veces superior al valor de corte. La paciente reúne por tanto, y con exceso, los criterios diagnósticos antes mencionados.
Dawson et al (18) realizaron un estudio en Portugal de ataxias hereditarias que tuvieran apraxia óculomotora entre sus manifestaciones clínicas. La apraxia óculomotriz que presentaba nuestra paciente es la que se describe en ese trabajo como característica de la ataxia-telangiectasia, presentando, además, otras dos ataxias hereditarias: la ataxia espinocerebelar tipo 2 y la ataxia recesiva con apraxia óculomotora descrita por Barbot et al (19). La disminución del tono muscular y de los reflejos osteotendinosos es consecuencia de la instalación progresiva de una neuropatía periférica, si bien en una edad algo menor a la descrita en la literatura (11,13).
Las infecciones respiratorias recidivantes causaron una alteración permanente del V/Q por daño alveolar que se tradujo en hipoxemia crónica evidenciada por el hipocratismo digital y la O2 dependencia. Estas infecciones ocurren por la alteración de la inmunidad celular y humoral que sufren estos enfermos y que se demostró en los resultados de laboratorio.
Sadighi et al(20) investigaron la presencia de hipergammaglobulinemia y gammopatías oligo y monoclonales en 90 pacientes con ataxia-telangiectasia. Un aumento aislado en IgM fue el hallazgo más común (23,3%), tal como se observó en la paciente. Por otra parte la disminución marcada de las otras clases de inmunoglobulinas es un hallazgo frecuente en estos pacientes y se encontraba también en el presente caso (11).
En el estudio inmunofenotípico no se encontraron linfopenia, inversión de la relación CD4/CD8, ni otras alteraciones descritas en la bibliografía, siendo normales todas las subpoblaciones estudiadas excepto en el aumento de los NK para los rangos referidos por Katevas et al(14), esto es ligeramente por encima de dos DS, mientras se encuentra por debajo del percentil 95 utilizado por Ikinciogullari et al (15) aunque la distribución es no normal; y en la notable disminución de los linfocitos T cooperadores “naive” (CD4+/CD45RA+), tanto para valores de referencia en adultos como en niños.
Schubert et al(21) estudiaron las deficiencias en los subtipos de células T CD4+ y CD8+ en ataxia-telangiectasia. Encontraron que los linfocitos CD45RA+ (“naive” o vírgenes – emigrantes tímicos tempranos) se encuentran fuertemente disminuidos, mientras que los NK (CD56+) están significativamente elevados en estos enfermos. Utilizando varios rangos de referencia pediátricos(14,15,22) quedan únicamente como claramente anómalos en esta paciente los bajos niveles de la población CD45RA+.
El análisis citogenético mostrará en los homocigotos y portadores alteraciones cromosómicas espontáneas con una elevada frecuencia(23). Dichas alteraciones serán de utilidad como un elemento confirmatorio más, o diagnósticas, en aquellos raros casos (menos del 5%), donde los niveles de alfa fetoproteína no se encuentran elevados. En nuestro caso dicho estudio no fue realizado.
La identificación de fragmentación del ADN post-irradiación ha permitido el diseño de técnicas de cribado para los heterocigotos de relativa sencillez(24). Por otra parte, la detección precisa de la alteración génica representa un gran desafío, derivado del alto número de modificaciones (mayores a 400 descritas) distribuidas en los 66 exones del gen ATM. Son, por tanto, atribución de escasos laboratorios en el mundo (25).
El interés creciente en detectar el estado de portadores (heterocigotos) va más allá del posible consejo genético en una enfermedad de muy baja frecuencia, para relacionarse con el riesgo aumentado a neoplasia en los portadores.
La ataxia-telangiectasia pertenece a un grupo de enfermedades colectivamente denominadas “síndromes de inestabilidad genómica” dentro de las cuales y aparte de la ataxia-telangiectasia, encontramos: el síndrome de Nijmegen, la anemia de Fanconi, el xeroderma pigmentoso, el síndrome de Bloom y otras cinco infrecuentes enfermedades.
Siendo los mecanismos diferentes en cada caso, todas ellas conllevan la incapacidad de detectar la lesión del ADN, transmitir el mensaje o finalmente, reparar el daño sufrido en el mismo (26). El presente caso puede servir de alerta y motivo de atención para estas enfermedades.
Agradecimientos
A la Dra. Cecilia Canessa por la realización del inmunofenotipado. Al Prof. Agdo. Dr. Luis Borche por la lectura crítica del manuscrito.
Referencias bibliográficas
1. Syllaba L, Henner K. Contribution a l’independence de l’athetose double idiopathique et congenitale. Rev Neurol 1926; 1: 541-62.
2. Louis-Bar D. Sur un syndrome progresiff comprenant des télangiectasies capillaries cutanées et conjontivales symétriques à disposition naevoîde et des troubles cérébelleux. Confin Neurol. 1941; 4: 33-42.
3. Boder E, Sedgwick RO. Ataxia-telangiectasia: a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia, and frequent pulmonary infection. Univ S Calif Med Bull 1959; 9: 15-27.
4. Rebollo MA. Ataxia-telangiectasia. Acta Neurol Latinoamer 1961; 7: 331-336.
5. Marcos J, García-Güelfi A, Maggi R, Sande MT. Ataxia-telangiectasia (enfermedad de Louis-Bar). Arch Ped Urug 1962; 33: 530-6.
6. Gatti RA. Ataxia-telangiectasia. In: Scriver CR, Beaudat AL, Sly WS. Metabolic and Molecular Basis of Inherited Diseases. 8ª ed. New York: McGraw-Hill, 2001: 705-32.
7. Sanal O, Lange E, Telatar M, Sobel E, Salazar-Novak J, Ersoy F, et al. Ataxia-telangiectasia : linkage analysis of chromosome 11q22-23 markers in Turkish families. (OMIM 208900) FASEB J 1992; 6: 2848-52. <http://www.pubmed.com> [consulta: 20 mar 2005]
8. Gatti RA, Berkel I, Border E. Localisation of the ataxia-telangiectasia gene to chromosome 11q22-23. Nature 1988; 336: 577-80.
9. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia-telangiectasia gene with a product similar to PI-3 Kinase. Science 1995; 268: 1749-53.
10. Elder M. Inmunodeficiencias de células T. Clin Pediatr Norteam 2001; 6: 1299-300.
11. Regueiro J, Porras O, Lavin M, Gatti R. Ataxia-telangiectasia: A primary immunodeficiency revisited. Clin Inmunol Alerg Norteam 2000; 20: 177-205.
12. Swift A, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991; 325: 1831-6.
13. Prats Viñas JM. Ataxias y síndrome cerebeloso. In: Fejerman N. Neurología pediátrica. 2ª ed. Buenos Aires: Médica Panamericana, 1997: 434-46.
14. Katevas P, Lembesopoulos C, Kamitaki C, Sakellariou D, Douna V. Establishment of normal ranges of lymphocite subpopulations in cord blood and inperpheral blood in infants and children. Haema 1999; 2: 145-151.
15. Ikinciogullari A, Kendirli T, Dogu F, Egin Y, Reisli I, Cin S, et al. Peripheral blood lymphocyte subsets in healthy Turkish children. Turk J Pediatr 2004; 46: 125-30.
16. Cabana MD, Crawford TO, Winkelstein JA, Christensen JR, Lederman HM. Consequences of a delayed diagnosis of ataxia-telangiectasia. Pediatrics 1998; 102: 98-100.
17. Waldman TA, McIntyre KR. Serum alpha-fetoprotein levels in patients with ataxia-telangiectasia. Lancet 1972; 2: 112.
18. Dawson D. Recessive ataxia with ocular motor apraxia. Arch Neurol 2001; 58 (2): 173-4.
19. Barbot C, Coutinho P, Chorao R, Ferreira C, Barros J, Fineza I, et al. Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch Neurol. 2001; 58(2): 201-205.
20. Sadighi A, Amir A, Phil D, Humphrey R, Winkelstein J, Loeb D, et al. Oligo-monoclonal gammophaty and hypergammaglobulinemia in ataxia-telangiectasia: a study of 90 patients. Medicine (Baltimore) 1999; 78 (6): 370-81.
21. Schubert R, Reichenbach J, Zielen S. Deficiencies in CD4+ and CD8 T cell subsets in ataxia telangiectasia. Clin Exp Immunol 2002; 129 (1): 125-32.
22. Comans-Bitter WM, Groot R, Beemd R, Neijens H, Hop W, Groeneveld K, et al. Immunophenotyping of blood lymphocytes in chilhood. J Pediatr 1997; 130: 388-93.
23. Aurias A, Dutrillaux B, Burit D, Lejeune J. High frequencies of inversions and translocations of chromosomes 7 and 14 in ataxia-telangiectasia. Mutat Res 1980; 69: 369-74.
24. Djuzonova CS, Schinder D, Stopper H, Hoehn H, Flentje M, Oppitz U. Identification of ataxia-telangiectasia heterozygotes, a cancer prone population using the single-cell electrophoresis (Commet) assay. Lab Invest 1999; 79: 699-705.
25. Castellvi-Bel S, Sheikhavandi S, Telatar M, Tai L-Q, Hwang M, Wang Z, et al. New mutations, polymorphisms, and rare variants in the ATM gene by a novel strategy. Hum Mutat 1999; 14: 156-62.
26. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature Rev Cancer. 2003; 3: 155-68.
Correspondencia : Dr. Marcos Delfino
Mateo Cortés 4970 C. Montevideo, Uruguay
E-mail: melidelfino@adinet.com.uy


{kind=link}