










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.77 no.1 Montevideo 2006
POSTER
Arch Pediatr Urug 2006; 77(1): 45-49
CAGGIANI M 1, JURADO R 2, PANDOLFO S 3, BELLINZONA G 4
1. Ex Prof. Adjunto de Pediatría. Clínica Pediátrica C. Jefe de la Policlínica de Enfermedades Colagenovasculares CHPR.Nefrólogo. Docente Honorario de la Facultad de Medicina de Montevideo. Universidad de la República.
2. Docente Honorario de la Facultad de Medicina de Montevideo. Universidad de la República. Médico Asistente de la Policlínica de Enfermedades Colagenovasculares del CHPR y Policlínica de Reumatología.
3. Residente de Pediatría. Clínica Pediátrica C. Prof. Dra. I. Rubio.
4. Prof. Adjunto de Pediatría. Clínica Pediátrica C. Prof. Dra. I. Rubio.
Policlínica de Enfermedades Colagenovasculares. Hospital Pereira Rossell. Facultad de Medicina.
Póster presentado en XXV Congreso Uruguayo de Pediatría, I Congreso Uruguayo de Neonatología, I Congreso Uruguayo de Enfermería Pediatría, 1ra. Jornada de Residentes en Pediatría del Cono Sur, año 2005
Resumen
Introducción: la dermatomiositis juvenil (DMJ) es una enfermedad mutisistémica del tejido conectivo, caracterizada por inflamación del músculo estriado y la piel con presencia de vasculitis como alteración anatomopatológica subyacente. El diagnóstico de la DMJ se basa en los criterios de Bohan y Peter propuestos en 1975: debilidad muscular proximal y simétrica de cintura escapular y pelviana, elevación de enzimas musculares: creatininfosfoquinasa, aldolasa, lactodeshidrogenasa y transaminasas, cambios electromiográficos con las características de miopatía y denervación. Biopsia muscular compatible con necrosis e inflamación, lesiones cutáneas características. Es imprescindible la presencia de rash cutáneo para realizar el diagnóstico. Este es seguro con cuatro criterios, probable con tres y posible con un criterio.
Objetivo: presentar ocho casos de DMJ atendidos en la Policlínica de Enfermedades colagenovasculares del CHPR en el período comprendido entre octubre de 2003 a agosto de 2005.
Material y métodos: se realizó un estudio descriptivo, retrospectivo de los casos de DMJ de la policlínica de Enfermedades colagenovasculares del CHPR registrados del 1º de octubre del 2003 a el 1º de agosto del 2005.
Resultados: 50% correspondieron al sexo femenino, la edad de presentación tuvo una media de ocho años. Con respecto a la clínica, presentaron manifestaciones cutáneas características, así como debilidad muscular y repercusión general 100%, manifestaciones osteoarticulares 87,5% , respiratorias 62,5% y digestivas 50%. En cuanto a la paraclínica, 100% presentaron enzimas musculares elevadas, 63% electromiograma característico, del total de biopsias musculares realizadas 40% fueron patológicas. El 100% recibió corticoides, sólo un paciente recibió monoterapia. 62,5% se encuentran en remisión total, 37,5% en remisión parcial.
Conclusiones: la distribución por sexos fue similar, el grupo etario con una media de presentación de 8 años coincide con la bibliografía. La mayoría de los pacientes (87,5%) cumplieron con cuatro de los criterios diagnósticos, sólo uno presentó tres criterios. Se destaca la presencia de manifestaciones multisistémicas, predominantemente respiratorias y digestivas. No fue necesario realizar a todos la biopsia muscular, salvo en aquellos casos que fue preciso para confirmar el diagnóstico. La biopsia muscular normal, no descartó el diagnóstico de DMJ ya que es una enfermedad de afectación focal. Todos recibieron corticoides siendo los fármacos de primera línea en esta enfermedad ya que disminuyen la morbimortalidad. Los fármacos de segunda línea se utilizaron en los casos de grave presentación inicial o falta de respuesta a la corticoterapia.
Summary
Introduction: JDM is a multisystemic connective tissue disease with the anatomic characteristics of muscular inflammation and vasculopathy. The diagnosis is based on Bohan and Peter´s criteria which are proximal and symmetrical scapular and pelvic muscular weakness; elevated muscle-derived enzymes (creatine kinase, aldolase, lactic acid dehydrogenase and transaminases); electromyographic changes; muscular biopsy that shows necrosis and inflammation and the presence of skin rash which is substantial.The diagnosis is certain with the four criteria present, probable with three and possible with the presence of 1.
Objective: to describe eight patients who were seen between October 2003 and August 2005 at the public hospital (Centro Hospitalario Pereira Rossell).
Material and methods: a descriptive, retrospective study of registered JDM cases was done from October 2003 until August 2005.
Results: half of the patients were female and the average presentation age was eight years old. 100% had cutaneous manifestations and muscle weakness; 87.5% had bone and joints symptoms; 62.5% had respiratory symptoms and 50% had digestive symptoms. The laboratory findings showed elevated serum enzymes in 100% of the cases, characteristic eletromyogram in 63% and 40% of the biopsies done were diagnostic. All patients were treated with corticoids. Disease remission is happening in 62,5% of the cases, partial remission in 37,5%.
Conclusions: the age prevalence were similar to international literature. All patients had skin rash and muscular manifestations. Muscular biopsy was realized in patients who needed confirmation. Normal biopsy did not discard the diagnosis due to the fact that it is an active focal disease. All patients were treated with corticoids. Second line drugs were used when the expected evolution did not happen or in cases of severe initial presentation.
Introducción
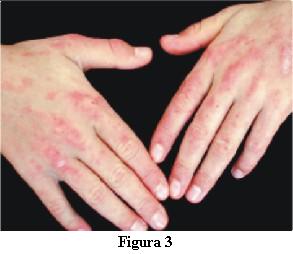
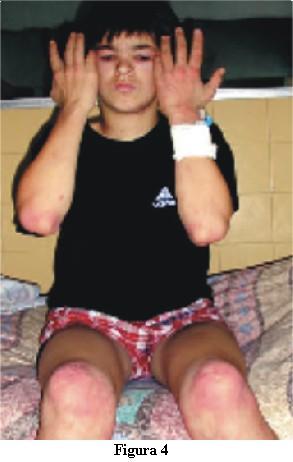
La dermatomiositis juvenil (DMJ) es una enfermedad mutisistémica del tejido conectivo, caracterizada por inflamación del músculo estriado y la piel con presencia de vasculitis como alteración anatomopatológica subyacente.
El diagnóstico de la DMJ se basa en los criterios de Bohan y Peter propuestos en 1975 :
1. Debilidad muscular proximal y simétrica de cintura escapular y pelviana.
2. Elevación de enzimas musculares: creatininfosfoquinasa, aldolasa, lactodeshidrogenasa y transaminasa.
3. Cambios electromiográficos: con las características de miopatía: ondas de baja amplitud, corta duración, polifásicas y denervación: fasiculaciones en reposo, descargas de alta frecuencia anómala.
4. Biopsia muscular con necrosis e inflamación.
5. Lesiones cutáneas características.
Es imprescindible la presencia de rash cutáneo para realizar el diagnóstico. Este es seguro con cuatro criterios, probable con tres y posible con un criterio.
Objetivo
Presentar ocho casos de DMJ atendidos en Policlínica de Enfermedades colagenovasculares del Centro Hospitalario Pereira Rossell en el período comprendido entre el 1º de octubre del 2003 a el 1º de agosto del 2005.
Material y método
Se realizó un estudio descriptivo, retrospectivo de los casos de DMJ registrados del 1º de octubre del 2003 al 1º de agosto del 2005.
Se analizaron sexo, edad, formas de presentación clínica, exámenes de laboratorio, tratamiento, evolución y complicaciones.
Resultados
Cuatro correspondieron al sexo femenino y cuatro al masculino.
La edad de presentación fue entre los 4 a 13 años de edad con una media de 8 años.
Las formas de presentación se muestran en la tabla 1.
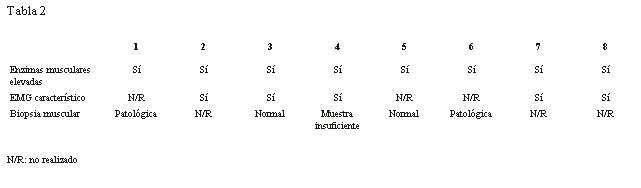
Los exámenes de laboratorio se muestran en la tabla 2.
Con respecto a la paraclínica presentaron:
1. 100% enzimas musculares elevadas.
2. 63% EMG característico.
3. 40% biopsias patológicas de las cinco realizadas.
Con respecto al tratamiento (figura 1) recibieron:
- Corticoides: 100%, 5/8 (62,5%) bolos de metilprednisolona.
- Inmunoglobulinas: 50%.
- Metrotexate: 87,5%.
- Ciclofosfamida o ciclosporina: 12,5%, respectivamente.

El seguimiento de los pacientes tuvo una media de 2 años y 4 meses.
Evolución (figura 2):
- Remisión sin tratamiento: 13%.
- Remisión con tratamiento: 49%.
- Remisión parcial: 38%.
- Recaída: un caso (12,5 %), luego de 6 años sin tratamiento. Actualmente en remisión.

Complicaciones:
- Cushing: 37,5%.
- Infecciosas: 62,5%:
- celulitis, abscesos;
- neumonía;
- sarna noruega.
Conclusiones
- La edad de presentación tuvo una media de 8 años coincidiendo con lo referido en la literatura.
- La distribución por sexo fue similar, no observándose el predominio en el sexo femenino.
- La mayoría de los pacientes (87,5%) cumplieron con cuatro de los criterios diagnósticos, sólo uno presentó tres criterios.
- Se destaca la presencia de manifestaciones sistémicas predominantemente respiratorias (62,5%) y digestivas (50%).
- La evolución con el tratamiento inmunosupresor fue favorable con alto porcentaje de remisiones y ausencia de mortalidad.
- Las complicaciones infecciosas se vieron en un 62,5% de los casos y el síndrome de Cushing en un 37,5%.


{kind=link}
{kind=link}
{kind=link}