










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.77 no.1 Montevideo 2006
PAUTAS
Arch Pediatr Urug 2006; 77(1): 39-42
Pauta de tratamiento del púrpura trombocitopénico inmune
Comité de Terapéutica del Centro Hospitalario Pereira Rossell (año 2003) 1
Servicio de Hemato-Oncología Pediátrica 2
2. Servicio de Hemato-Oncología Pediátrica: Dr. Gustavo Dufort, Dr. Agustín Dabezies, Dr. Luis Castillo.
Introducción
El púrpura trombocitopénico inmune (PTI) es una enfermedad relativamente poco frecuente (incidencia ~4/100.000 niños) que se caracteriza por un descenso adquirido de las plaquetas circulantes. Es una enfermedad donde el niño ha formado anticuerpos contra sus propias plaquetas, lo que resulta en un aumento de la destrucción de las plaquetas circulantes. La patogenia del PTI no se comprende completamente. Muchos episodios de PTI ocurren después de una infección viral o una inmunización. Las consecuencias clínicas incluyen equimosis, sangrados mucosos y petequias en un niño que por lo demás está sano. En los niños el PTI es generalmente una enfermedad benigna y autolimitada, con recuperación completa y permanente. La complicación más temida es el sangrado intracraneano que afortunadamente sólo ocurre raramente (0,1% de los casos de PTI agudo). Un problema adicional es que, en aproximadamente el 20-30% de los casos, la trombocitopenia no se resuelve después del inicio del cuadro clínico. Si el PTI continúa por más de 6 meses, esta condición se define como PTI crónico, lo que implica la persistencia de riesgo de vida por sangrado.
Mientras la comunidad médica ha ganado experiencia en el manejo del PTI, no hay consenso en cual es el mejor tratamiento. La tendencia de la evolución natural de la enfermedad a su curación espontánea, frente al riesgo hemorrágico en pacientes con trombocitopenia grave, es la causa de actitudes terapéuticas diversas. Los estudios disponibles no han permitido dilucidar las principales interrogantes: ¿Se necesita realmente tratamiento para aumentar el número de plaquetas hasta una cifra de bajo riesgo de sangrado? ¿Es necesario tratar a todos los pacientes con PTI? ¿Cuál es el objetivo terapéutico? ¿Cuáles son los beneficios del tratamiento? Entre los recursos terapéuticos disponibles, corticoides, inmunoglobulina intravenosa (IGIV) e inmunoglobulina anti-D (Ig Anti-D), ¿cuál es la mejor opción?
En el marco del programa de “Uso racional del medicamento” en el Centro Hospitalario Pereira Rossell, impulsado por el Comité de Terapéutica del Hospital Pediátrico y por la Unidad de Farmacología Clínica, se realizó una revisión bibliográfica del tratamiento del PTI.
El objetivo de la revisión fue analizar la evidencia disponible con relación a la eficacia y seguridad del tratamiento de esta patología.
Se consultaron las bases de datos Medline y Cochrane en los últimos 10 años.
Los artículos disponibles se analizaron en forma crítica.
Para determinar los niveles de evidencia se utilizaron los criterios publicados por la Sociedad Americana de Hematología (tabla 1).

Resultados de la revisión
1. Objetivo terapéutico
En la mayoría de los estudios la eficacia del tratamiento es valorada a través de la tasa de aumento del recuento plaquetario, sin relacionarla con la severidad del sangrado o la mortalidad. El recuento plaquetario es una variable subrogada que se correlaciona con la clínica y por tanto tiene las siguientes limitaciones:
- La asociación entre número de plaquetas y hemorragia ha sido demostrada para pacientes con plaquetopenias diferentes al PTI.
- El beneficio terapéutico puede ser debido en parte a efectos no relacionados con el recuento plaquetario.
- Los niños con PTI tienden a presentar menor número de hemorragias y más escasas con cualquier número de plaquetas comparados con niños con leucemia, anemia aplásica o tratamiento quimioterápico (III, IV, V). En el caso del PTI las plaquetas son jóvenes, tienen mayor adhesividad y la reserva medular es excelente. Por lo tanto la mayoría de los autores señalan que el principal objetivo terapéutico es acelerar la recuperación del recuento plaquetario.
2. Hospitalización
No se dispone de estudios evaluando este aspecto del tratamiento. La mayoría de los autores recomienda hospitalizar a los niños con un número de plaquetas £ 20.000/mm3 y sangrado mucoso.
3. Observación (sin tratamiento farmacológico)
Entre 1992 y 1999 Dickerhoff R analiza en forma retrospectiva la evolución de 55 niños con PTI sin tratamiento. En ese estudio, 37 de los 55 pacientes tuvieron recuentos plaquetarios < 10.000/mm3, la mayoría (n = 27) no presentó sangrado mucoso activo. Cinco pacientes con sangrado mucoso tenían recuentos plaquetarios entre 10.000 y 20.000/mm3. Ningún paciente presentó sangrado de riesgo vital ni falleció (V).
Se dispone de ensayos clínicos controlados comparando la eficacia de IGIV, corticoides orales y placebo. En estos estudios no se demostró diferencia estadísticamente significativa en el tiempo en alcanzar niveles plaquetarios mayores de 50.000/mm3 entre los niños que recibieron IGIV y esteroides, pero sí la hubo entre el grupo sin tratamiento y con tratamiento (2, 4 y 6 días respectivamente). En estos estudios ningún paciente presentó hemorragia grave ni falleció (I).
Algunos autores señalan que el manejo de estos pacientes sin tratamiento específico evita la hospitalización innecesaria, los efectos adversos y reduce los costos.
4. Glucocorticoides versus IGIV
Se han realizado varios ensayos clínicos comparando la eficacia de los glucocorticoides orales en diferentes dosis con IGIV (Blanchette V 1993 y 1994; Rosthoj S 1996; Duru F 2002). Como se señaló anteriormente en todos los estudios la variable principal de medida es la velocidad de aumento del recuento plaquetario. En la mayoría de ellos la respuesta es más rápida en los grupos tratados con IGIV, diferencia uno a dos días (I). El proceso de recuperación espontáneo es acelerado con la administración de glucocorticoides o IGIV, pero tal reacción es transitoria y no brinda protección contra la hemorragia mortal ni previene la evolución a la cronicidad (III, IV, V).
En las guías alemana y británica para el manejo del PTI se recomienda iniciar el tratamiento en base a la valoración de la condición del paciente (magnitud del sangrado) más que en el recuento plaquetario. En estas guías no se recomienda tratamiento específico en pacientes con recuentos plaquetarios < 10.000/mm3 sin sangrado activo o con recuentos plaquetarios > 20.000/mm3 con sangrado mucoso. Se reserva la administración de IGIV para pacientes con sangrado de riesgo vital (V).
Ambos tratamientos se acompañan de efectos adversos. En los grupos tratados con IGIV se han notificado náuseas, vómitos, cefaleas y fiebre. La meningitis aséptica es una reacción adversa rara. El efecto adverso más frecuente comunicado durante la administración de glucocorticoides es el aumento de peso. Otros efectos como osteopenia, hipertensión arterial y aumento de la glucemia dependen de la dosis y duración de la corticoterapia (I).
5. Inmunoglobulina anti-D
La Ig anti-D fue recién aprobada por la FDA para su uso en el PTI en año 1995, e incluida en las recomendaciones de la Sociedad Americana de Hematología en el año 1996. Existe experiencia clínica acumulada que indica su efectividad comparable a la IGIV y los corticoides. Su uso está limitado a pacientes Rh positivos. Es un tratamiento seguro, de fácil administración y menor costo que la IGIV. El único efecto adverso importante es la hemólisis aloinmune.
Guía para el diagnóstico y tratamiento inicial
Basados en los niveles de evidencia disponibles y en las recomendaciones del Grupo de trabajo de la Sociedad Española de Hematología Pediátrica y de la Sociedad Americana de Hematología, el Comité de Terapéutica del Hospital Pediátrico y el Servicio de Hemato-Oncología Pediátrica del Centro Hospitalario Pereira Rossell, elaboraron la siguiente guía para diagnóstico y tratamiento del PTI.
A. Diagnóstico
El diagnóstico de PTI se basa en:
- Una historia clínica y examen físico que excluyen otras causas de trombocitopenia.
- El hemograma con estudio de frotis de sangre periférica es esencial. A excepción de la trombocitopenia el resto de los valores deben ser normales o si están alterados debe ser por causas fácilmente explicables, como por ejemplo anemia por déficit de hierro o talasemia menor. La morfología de los glóbulos blancos debe ser normal aunque algunos pacientes pueden tener linfocitos atípicos o eosinofilia.
- Otros exámenes de laboratorio no están indicados de realizar para el diagnóstico de PTI. Sólo se recomienda testar el VIH en pacientes con factores de riesgo.
- El estudio de médula ósea no se realizará para establecer el diagnóstico previo al inicio del tratamiento. Se realizará sólo en aquellos pacientes que no respondan al tratamiento inicial.
B. Tratamiento
1. Recomendaciones generales
- Ingreso hospitalario (fase aguda con recuento plaquetario <50.000/mm3).
- Consulta con hematólogo pediatra.
- No administrar fármacos por vía intramuscular.
- Contraindicado el uso de ácido acetilsalicílico y derivados, y otros fármacos que puedan alterar la agregación plaquetaria (antihistamínicos, AINES).
- Escolarización normal si tiene recuento plaquetario estable superior a 50.000/mm3.
- Deportes: restricción de la actividad física hasta resolución de la enfermedad.
- Seguimiento en Policlínica de Hematología Pediátrica.
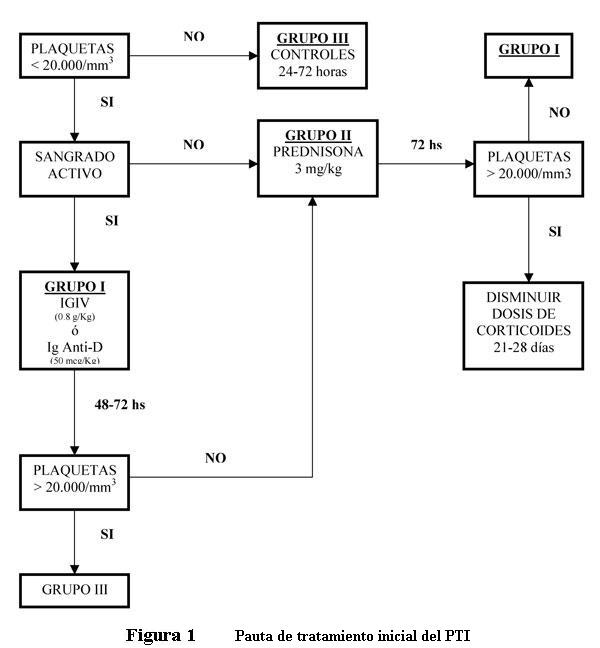
2. Clasificación de los pacientes
- Grupo I: niños con menos de 20.000 plaquetas por mm3 y/o sangrado activo en mucosas (epistaxis que no cede con taponamiento, hematuria macroscópica, gingivorragia, hemorragia gastrointestinal, menorragia).
- Grupo II: niños con menos de 20.000 plaquetas por mm3 sin sangrado activo en mucosas.
- Grupo III: niños con recuento plaquetario > 20.000/mm3.
3. Esquema de tratamiento
- Grupo I: IGIV o Ig Anti-D (solo pacientes Rh+) dosis única. Valoración clínica a las 24 horas: si persiste el sangrado activo se añaden corticoides; si desaparece la clínica, recuento plaquetario a las 48 horas; si es menor a 20.000 elementos/mm3 pasa a la pauta de tratamiento del Grupo II;; si desaparece la clínica y el recuento plaquetario es superior a 20.000 elementos/mm3 pasa a la pauta de tratamiento del Grupo III.
- Grupo II: corticoides vía oral. Nueva valoración a las 72 horas: si persiste recuento plaquetario inferior a 20.000 elementos//mm3 y/o clínica de sangrado activo en mucosas, se administra una dosis de IGIV o Ig Anti-D (Grupo I); si supera las 20.000 plaquetas por mm3 se continúa tratamiento con corticoides con nuevo control a los siete días.
- Grupo III: conducta expectante durante 2 semanas. Controles periódicos cada 24 a 72 horas. Si a la segunda semana no hay aumento del recuento plaquetario valorar inicio de tratamiento con corticoides.
- Tratamiento de emergencia: el manejo del paciente con sangrado que determine riesgo de vida justifica el uso de varios regímenes combinados: IGIV, altas dosis de corticoides (metilprednisolona 30 mg/kg por 3 días) y transfusión de plaquetas.
- Tratamiento en el recién nacido: en el RN con PTI o hijo de madre con PTI se debe considerar el riesgo de hemorragia intracraneana (HIC) con recuentos plaquetarios inferiores a 50.000 por mm3. De constatarse HIC realizar tratamiento de emergencia. RN sin HIC con recuento plaquetario menor de 30.000/mm3 realizar IGIV.
4. Dosis recomendadas
- IGIV: 0,8 g/kg, dosis única.
- Ig anti-D: 50 mg/kg, dosis única.
- Prednisona 3 mg/k/día v/o repartida en 2 o 3 dosis la primera semana; segunda semana 1 mg/kg, si el recuento plaquetario es mayor de 40.000 por mm3; tercera y cuarta semana descenso progresivo hasta supresión.
Figura 1: Pauta de tratamiento inicial del PTI
Referencias bibliográficas
1. George J, Woolf S, Raskob G, Wasser J, Aledort L, Ballem P, et al. Idiopathic Thrombocytopenic Purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood 1996; 88(1): 3-40.
2. Grupo de trabajo de la Sociedad Española de Hematología Pediátrica. Protocolo de estudio y tratamiento de la púrpura trombocitopénica inmune. An Esp Pediatr 1996; 44: 623-31.
3. Sorian Guillén I, Sánchez Bayle M, Mencia Bartolomé S, Martínez Martín C. Púrpura trombocitopénica inmune: revisión de 113 casos. Rev Esp Pediatr 2000; 56(6): 489-93.
4. Dickerhoff R, von Ruecker A. The clinical course of inmune thrombocytopenic purpura in children who did not receive intravenous immunoglobulins or sustained prednisone treatment. J Pediatr 2000; 137 (5) 629-32.
5. Blanchette V, Imbach P, Andrew M, Adams M, McMillan J, Wang E, et al. Randomized trial of intravenous immunoglobulin G, intravenous anti-D, and oral prednisone in acute immune thrombocytopenic purpura. Lancet 1994; 334(10): 703-7.
6. Blanchette V, Chir B, Luke B, Andrew M, Sommerville-Nielsen S, Barnard D, et al. A prospective, randomized trial of high-dose intravenous immune globulin G therapy, oral prednisone therapy, and no therapy in childhood acute immune thrombocytopenic purpura. J Pediatr 1993; 123(6): 989-95.
7. George JN. Editorial: Initial management of immune thrombocytopenic purpura in children: Is supportive counseling without therapeutic intervention sufficient? J Pediatr 2000; 137(5): 598-600.
8. Medeiros D, Buchanan G. Major hemorrhage in children with idiopathic thrombocytopenic purpura: immediate response to therapy and long-term outcome. J Pediatr 1998; 133(3): 334-39.
9. Rosthoj S, Nielsen S, Karup-Pedersen F. Randomized trial comparing intravenous immunoglobulin with methylprednisolone plus therapy in acute idiopathic thrombocytopenic purpura. Acta Pediatr Int J Pediatr 1996; 85(8): 910-15.
10. Duru F, Fisgin T, Yarali N, Kara A. Clinical course of children with immune thrombocytopenic purpura treated with intravenous immunoglobulin G or megadose methilprednisolone or observed without therapy. Pediatr Hematol Oncol 2002; 19(4): 219-25.
11. Medeiros D, Buchanan G. Controversias actuales en el tratamiento de la púrpura trombocitopénica idiomática en niños. Clin Pediatr Norteam 1996; 3: 707-21.
12. Lilleyman JS. Intracranial hemorrage in idiopathic thrombocytopenic purpura. Arch Dis Child 1994; 71: 251-53.
13. Cines D, Blanchette V. Immune Thrombocytopenic Purpura. N Engl J Med 2002; 346(13): 995-1008.
14. Sandler SG. Intravenous Rh immune globulin for treating immune thrombocytopenic purpura. Curr Opin Hematol 2001; 8: 417-420.


{kind=link}