










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.76 no.4 Montevideo dic. 2005
CASO CLÍNICO
Arch Pediatr Urug 2005; 76(4): 305-311
Dermatomiositis juvenil.
A propósito de un caso con grave compromiso multisistémico
DRAS. SOLEDAD PANDOLFO 1, MARINA CAGGIANI 2, IVONNE RUBIO 3
1. Residente de Pediatría. Clínica Pediátrica C. Hospital Pereira Rossell.
2. Ex Profesora Adjunta de Clínica Pediátrica C. Docente Honoraria de Facultad de Medicina. Universidad de la República. Montevideo, Uruguay.
3. Profesora de Clínica Pediátrica C. Facultad de Medicina. Universidad de la República. Montevideo, Uruguay.
Autorizada la publicación de imágenes por el paciente y su madre.
Fecha recibido: 16 de setiembre 2005.
Fecha aprobado. 13 de diciembre 2005
Resumen
Se presenta un caso de dermatomiositis juvenil de presentación y evolución inhabitual en un paciente de 14 años de edad, que ingresa por una historia de 20 días de evolución caracterizada por manifestaciones cutáneas y musculares, constatándose al examen físico eritema en heliotropo, pápulas de Gottron y debilidad muscular proximal y simétrica.
Las enzimas musculares elevadas y el electromiograma característico completaron el diagnóstico.
Se destacó el grave compromiso respiratorio que obligó a la asistencia respiratoria mecánica, así como el toque multisistémico neurológico, digestivo y hematológico.
La inmunosupresión enérgica con corticoides y ciclofosfamida intravenosa, y el uso de altas dosis de inmunoglobulina intravenosa permitieron una evolución favorable manteniéndose en remisión luego de un año y medio con corticoides a baja dosis y metrotexate.
Palabras clave: DERMATOMIOSITIS-diagnóstico
DERMATOMIOSITIS-terapia
DERMATOMIOSITIS-complicaciones
Summary
A fourteen year old boy with an unusual presentation of juvenile dermatomyositis (JDM) is described.
The illness was characterized by cutaneous manifestations like heliotrope eyelids, Gottron papules and proximal muscle weakness.
Elevated serum levels of muscle-derived enzymes and the characteristic electromyogram completed the diagnosis of JDM.
Respiratory assistance was needed due to the severe respiratory failure. Multisystemic failure involving the central nervous system, gastrointestinal tract and hematopoyetic system occurred.
Aggressive immunosupression with corticosteroids, ciclophosphamide and intravenous immunoglobulin leaded to a favorable outcome. The disease has been in remission for 18 months with low doses of corticosteroids and methotrexate.
Key words: DERMATOMYOSITIS-diagnosis
DERMATOMYOSITIS-therapy
DERMATOMYOSITIS-complications
Introducción
La dermatomiositis juvenil (DMJ) es una enfermedad mutisistémica del tejido conectivo, caracterizada por inflamación del músculo estriado y la piel con presencia de vasculitis como alteración anatomopatológica subyacente. Cursa con debilidad muscular simétrica a predominio proximal como manifestación más frecuente, aunque es heterogénea en su forma de presentación así como en su evolución (1-4). Fue descrita por primera vez por Unverricht en 1900, en el Uruguay en 1949 por Plá y colaboradores. En pediatría la primera comunicación es en 1969 por Fabius y García (5).
La enfermedad tiene características propias en la infancia que la distinguen de la del adulto, como una tendencia mayor a la incapacidad física por atrofia muscular, mayor frecuencia de ulceraciones y perforación del tracto digestivo debido a vasculitis severa.
Su etiología es desconocida. Los estudios actuales sugieren una patogenia autoinmune y una predisposición genética asociándose a ciertos antígenos de histocompatibilidad: B8, DR3, DQA 1 y otros. Se forman anticuerpos que reaccionan frente a los capilares dentro del músculo ocasionando una vasculitis con isquemia y lesión de las células del mismo. Esta lesión muscular se traduce con la elevación de las enzimas musculares: creatininfosfoquinasa (CPK), lactatodeshidrogenasa (LDH), alanino aminotransferasa (TGP), aspartato aminotransferasa (TGO) y aldolasa. La CPK es el marcador más sensible (1,6-8).
No existen hasta el momento anticuerpos patognomónicos de dermatomiositis (2). Se han encontrado en algunos casos, poco frecuentes en niños, anticuerpos antisintetasa (antiamino-acetil-t RNA sintetasa: anti Jo-1) de los cuales se han descrito cinco. El cuadro clínico asociado a estos anticuerpos es una miositis más resistente al tratamiento, con enfermedad intersticial pulmonar (50-70%), artritis (90%) y fenómeno de Raynaud (60%). Otro anticuerpo detectado es el anti-PM/Scl. Éste se encuentra en un síndrome de solapamiento conocido como escleromiositis que asocia síntomas cutáneos de esclerodermia (2).
La DMJ es una enfermedad episódica, de aparición estacional, siendo más frecuente en primavera y verano, coincidiendo con la mayor incidencia de infecciones virales o bacterianas (1,4,5,9).
Se han implicado como desencadenantes de la enfermedad agentes infecciosos como enterovirus (coxsackievirus B), Toxoplasma gondii, estreptococo del grupo A, herpes simple, hepatitis A, influenzae A y B, parainfluenzae 1 y 3, micoplasma, sarampión, citomegalovirus y HIV (1,9). Se cree que existe una similitud molecular entre la miosina del músculo del estriado y un componente del agente infeccioso. Otro mecanismo propuesto es la participación de componentes bacterianos o virales como superantígenos.
Es una enfermedad poco frecuente, su verdadera incidencia y prevalencia es difícil de precisar debido a la rareza de la misma. La incidencia seria de 1 a 9 casos / millón de niños menores de 16 años al año (1,2,3,8). Si bien es poco frecuente el 16- 20% de todas las DM se inician en la niñez.
La edad de aparición es bimodal con un pico a los 5 a 15 años de edad y otro pico a los 45 a 54 años con predominio en el sexo femenino (1,3). En la bibliografía el caso clínico de menor edad corresponde a un niño de 2 años de edad (10).
Con respecto al inicio y evolución existen dos formas: aguda en un tercio de los casos en la que presenta un inicio y evolución rápida y la segunda de inicio y evolución lenta instalándose en el curso de 2 o 3 meses (1).
Objetivo
- Describir un caso clínico de DMJ con presentación y evolución poco frecuente destacándose la severidad del compromiso multisistémico.
Caso clínico
Adolescente de 14 años, sexo masculino, sin antecedentes personales ni familiares a destacar.
Motivo de ingreso: erupción cutánea y debilidad muscular.
Enfermedad actual: comienza 20 días previos al ingreso con fiebre de 38°C axilar que cede en 72 horas, anorexia y adelgazamiento. A las 24 horas agrega lesiones maculopapulosas eritematosas a nivel de cara, manos y miembros inferiores. Concomitantemente mialgias y debilidad muscular en los cuatro miembros, simétrica y a predominio proximal. No presenta alteraciones del tránsito digestivo y urinario.
Examen físico: paciente lúcido, con regular estado general, apirético. Peso: 42.500 g (P: 10-20). Talla: 150 cm (P: 70–80).
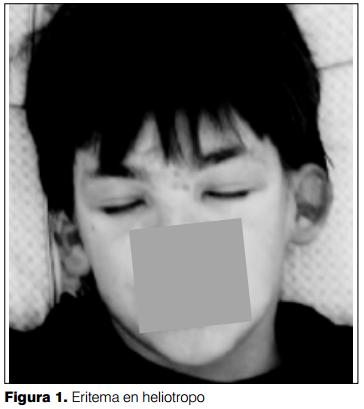
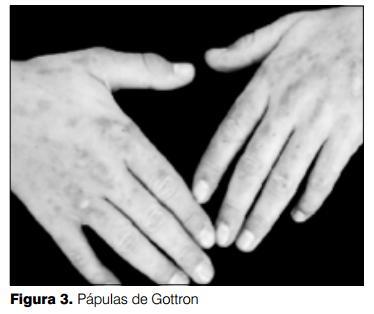
A nivel de la piel, se destaca la existencia de eritema y edema de ambos párpados de coloración violácea: eritema en heliotropo (figura 1, 2 y 3), lesiones maculopapulosas eritematosas en frente y axilas; en regiones extensoras de articulaciones de codo y rodillas placas psoriasiformes (figura 4). Sobre articulaciones metacarpo e interfalángicas de manos presenta placas eritematodescamativas con las características de pápulas de Gottron (figura 3 y 4).
A nivel linfoganglionar se encuentran microadenias carotídeas y axilares.
En el examen osteoarticular se observa dolor a la movilización pasiva y activa de las articulaciones de rodillas y hombros.
En el sector psiconeuromuscular, no presenta rigidez de nuca, pares craneanos sin alteraciones, a nivel espinal se destaca disminución de fuerzas y mialgias a predominio proximal de los cuatro miembros. Dificultad para elevar los brazos. Mingazzini y Gowers positivo. Reflejos osteotendinosos sin alteraciones. Sensibilidad normal. Marcha: fatiga al caminar unos pocos metros.
El examen abdominal y de fosas lumbares es normal.
A nivel pleuropulmonar presenta buena entrada de aire bilateral sin estertores. Cardiovascular: ritmo regular de 96 cpm, ruidos bien golpeados, sin soplos. Pulsos llenos. Presión arterial 80/50 mmHg.
Exámenes de laboratorio: enzimograma muscular: TGO 587 (N: 0-40 U/l), TGP 258 (N: 10-30 U/l), LDH 1860 (N:50-120) y CPK 5893 (N:24-125).
Electromiograma: perfil miopático en los cuatro miembros a predominio proximal.


Hemograma: glóbulos blancos 5.600 elementos/mm3, hemoglobina: 13 g/dl, plaquetas 71.000 elementos/mm3.
Velocidad de eritrosedimentación: 14 mm.
Fondo de ojo: exudados algodonosos en retina y engrosamiento venoso.
Radiografía de tórax: normal.
Electrocardiograma y ecocardiograma: normal.
Capilaroscopía: asas capilares dilatadas aisladas y en acúmulos.
Esofagogastroduodeno: normal.
Función renal y examen de orina: normal.
Funcional hepático: TGO 587, TGP 258, gamaglutamiltranspepetidasa: 36 (N: <50), colinestarasa 5.200 (N: 5.400–13.200).
Proteinograma electroforético: normal.
Serología para CMV: IgM positiva 1.350.
HIV negativo.
Evolución y tratamiento
Realizado el diagnóstico de DMJ se inició el tratamiento. Dado que presentó serología positiva para citomegalovirus se decide comenzar con fármacos de segunda línea como la gammaglobulina intravenosa a razón de 1 g/kg/día hasta obtener el resultado de la PCR para citomegalovirus.
Como tratamiento para el citomegalovirus se realizó ganciclovir intravenoso a razón de 10 mg/kg/día que se suspendió al obtener la PCR negativa.
Presentó buena evolución con mejoría de las lesiones cutáneas y recuperación progresiva de las fuerzas musculares.
Al séptimo día de internación instaló un anasarca. El proteinograma electroforético mostró una hipoproteinemia de 4,92 g% y una hipoalbuminemia de 0,44 g%, examen de orina: sin proteinuria, función renal: azoemia 0,56 g/l y creatininemia de 0,63 mg/dl, ionograma: Na 106 meq/l, K 5,7 meq/l. La ecografía de abdomen reveló una discreta ascitis en cavidad libre y la radiografía de tórax no mostró cardiomegalia ni derrame pleural. La dosificación de alfa 1 antitripsina en materias fecales fue de 6,50 mg/g (N: 1,5 mg/g de heces).
Se trasladó a la unidad de reanimación del Departamento de Emergencia (URE) dada la hiponatremia severa. Se repuso por fórmula con NaCl al 3% lento, se inició furosemide 1 a 2 mg/kg/dosis cada 6 horas, albúmina 0,5 g/kg i/v. Al recibir el PCR negativo para citomegalovirus se comenzó con bolos de metilprednisolona de 1 g/día durante tres días consecutivos.
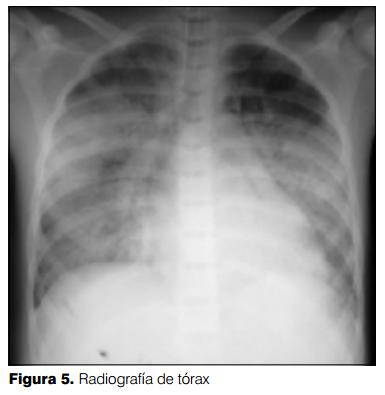
A las 24 horas de su traslado a la URE instaló depresión de conciencia, alucinaciones visuales e insuficiencia respiratoria, se solicitó radiografía de tórax (figura 5) que mostró una opacidad inhomogénea bilateral. Se trasladó a cuidados intensivos donde se realizó intubación orotraqueal, constatándose sangre en la vía respiratoria alta. Se inicia asistencia ventilatoria mecánica (AVM). Agregó a nivel digestivo enterorragia escasa sin repercusión hemodinámica. La tomografía de cráneo mostró leve dilatación ventricular sin alteraciones a nivel del parénquima cerebral. Ecocardiograma: leve hipertrofia del ventrículo izquierdo con una fracción de eyección del ventrículo izquierdo normal.
Se prosiguió con gammaglobulina intravenosa a dosis de 1 g/kg/día durante cinco días y se inició vista la radiografía de tórax, previo a la obtención de los cultivos tratamiento con vancomicina 40 mg/kg/día más ceftriazona 100 mg/kg/día. El cultivo de secreciones orotraqueales desarrolló Staphylococcus aureus meticilino resistente.
A nivel nutricional presentó una rápida disminución de las masas musculares y pérdida del tejido adiposo hasta llegar a la caquexia con pérdida de 15 kg del peso corporal con respecto al peso del ingreso, siendo necesaria la nutrición con preparados semielementales.
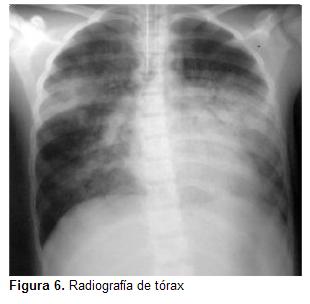
Al no mostrar mejoría evolutiva a las 48 horas (figura 6) de ingresar a UCIN, se realizó un bolo de ciclofosfamida de 700 mg y se mantuvo corticoterapia intravenosa con hidrocortisona 20 mg/kg/día.
Luego de 34 días de permanecer en cuidados intensivos mostró mejoría a nivel respiratorio (figura 7), se suspendió AVM, se mantuvo oxigenación con cánula nasal a 2 l/min por 48 horas con buena saturación, por lo que se trasladó a sala.
Al reingreso a sala el paciente presentó mal estado nutricional con mejoría de las lesiones cutáneas y de las fuerzas musculares, persistencia de las alucinaciones visuales y auditivas.
Desde el punto de vista terapéutico se indicó: dieta hiposódica e hipercalórica, prednisona a 2 mg/kg/día, metrotexate a 10 mg/m2/semana, calcio, vitamina D, omeprazol y ácido fólico.
Luego de la remisión de la signología clínica y la normalización de las enzimas musculares se comenzó un descenso gradual de la prednisona.
Se concedió el alta a los dos meses de internación con un peso estacionario de 32 kg, recuperación de las fuerzas musculares, remisión de las lesiones cutáneas, persistiendo con episodios de alucinaciones visuales y auditivas.
Al año y medio del comienzo de su enfermedad se encuentra con un excelente estado general y calidad de vida, concurre al liceo, practica deportes, el peso es adecuado para la edad, no presenta lesiones cutáneas, recuperó totalmente las fuerzas musculares, mantiene una CPK normal 90 U/l. Su tratamiento actual es: prednisona a dosis de 5 mg/día, calcio y vitamina D a iguales dosis, metrotexate a 15 mg semanal. No presentó complicaciones por su tratamiento corticoideo ni efectos secundarios del metrotexate.
El paciente es controlado desde el comienzo de la enfermedad en la Policlínica de Enfermedades Colagenovasculares del Centro Hospitalario Pereira Rossell.
Discusión
Se presentó el caso clínico de un adolescente de 14 años con una historia de 20 días de evolución caracterizada por lesiones cutáneas patognomónicas como el eritema en heliotropo y pápulas de Gottron, asociadas a debilidad muscular de los cuatro miembros, bilateral, simétrica y a predominio proximal, con aumento de las enzimas musculares y un electromiograma con un patrón miopático por lo que hacemos diagnóstico de dermatomiositis juvenil (DMJ).
El diagnóstico de la DMJ se basa en los criterios de Bohan y Peter propuestos en 1975 (1-3,8,11), que tiene una sensibilidad de 45-92% y una especificidad del 93%:
1. Debilidad muscular proximal y simétrica de cintura escapular y pelviana.
2. Elevación de enzimas musculares: creatininfosfoquinasa, aldolasa, lactodeshidrogenasa y transaminasas.
3. Cambios electromiográficos, con las características de miopatía: ondas de baja amplitud, corta duración, polifásicas y de denervación, fasiculaciones en reposo, descargas de alta frecuencia anómalas.
4. Biopsia muscular compatible con necrosis y miositis inflamatoria.
5. Lesiones cutáneas características.
Es imprescindible la presencia de rash cutáneo para realizar el diagnóstico que es seguro con cuatro criterios, probable con tres y posible con un criterio (1). Otros autores realizan el diagnóstico con el rash cutáneo asociado a otros dos criterios (8).
En este paciente no fue necesaria la realización de la biopsia muscular, ya que cumplió con cuatro criterios de la clasificación. Algunos autores la consideran innecesaria en estos casos dado que una biopsia muscular normal no excluye el diagnóstico por ser una enfermedad de afectación focal (1,8). Estaría indicado en casos de dudas diagnósticas o para excluir entidades como la miositis por cuerpos de inclusión en casos refractarios al tratamiento.
La biopsia muscular debe hacerse en músculos afectados pero no atrofiados. La resonancia nuclear magnética (RNM) puede realizarse para seleccionar el área afectada y disminuir las posibilidades de una biopsia muscular no informativa. Debe obtenerse una muestra generosa dado la afectación focal del músculo. No debe efectuarse en sectores en que se realizó el electromiograma.
El caso que hemos presentado se incluye en el tipo 4 de la clasificación de poliomiositis-dermatomiositis descrita en el año 1974 por Bohan y Peter (1,8).
- Tipo 1: poliomiositis.
- Tipo 2: dermatomiositis.
- Tipo 3: miositis con malignidad.
- Tipo 4: dermatomiositis juvenil.
- Tipo 5: miositis con otras colagenopatías.
- Tipo 6: miositis con cuerpos de inclusión.
La dermatomiositis juvenil tiene como base una vasculitis inmunológica que afecta no sólo el músculo sino que compromete a otros órganos (1,6,8). En este caso observamos compromiso neurológico, dado por la presencia de retinitis propia de esta enfermedad producida por vasculitis. La misma ocasiona focos de degeneración de las fibras nerviosas retinianas, dando lugar a la presencia de exudados algodonosos a nivel del fondo de ojo como en este caso. En nuestro paciente llevó a una disminución de la agudeza visual presentado resolución espontánea. En otros casos puede evolucionar a la ceguera (1,8).
La presencia de alucinaciones visuales y auditivas obedecería a la presencia de vasculitis a nivel cerebral destacándose la baja frecuencia del compromiso del sistema nervioso central (1,8).
El compromiso pulmonar grave sería secundario a una enfermedad pulmonar intersticial propia de la dermatomiositis. Ésta es una afectación fatal en muchas oportunidades. Se solicitó dosificación de anticuerpos anti –Jo que pueden ser marcadores de enfermedad pulmonar intersticial en la DMJ aunque su sensibilidad es baja en niños, en el adulto son positivos en un 50% de los casos. Por este motivo se está buscando otro marcador que sea más sensible y específico como la interleuquina 6 producida por los neumocitos tipo 2 y las células epiteliales cuando están en reparación y actúan como quimiotácticos para los fibroblastos (12).
La hemorragia pulmonar causada por la vasculitis a ese nivel, la infección facilitada por la inmunodepresión y la debilidad de la musculatura del tórax que favorece la aspiración se plantearon como causas asociadas de la insuficiencia respiratoria severa de este enfermo.
Probablemente se haya asociado el edema pulmonar facilitado por la reposición con NaCl hipertónico que reparó la hiponatremia severa que tuvo el paciente.
En el sistema cardiovascular el ecocardiograma pone de manifiesto alteraciones anatómicas como la hipertrofia leve del ventrículo izquierdo mientras que el ritmo cardíaco es normal. Las manifestaciones más frecuentes descritas de la vasculitis a nivel cardiovascular son los trastornos de la conducción y la pericarditis; la miocarditis es muy rara (1). En este caso no se observaron elementos clínicos, electrocardiográficos ni ecocardográficos de miocarditis, pericarditis ni trastornos del ritmo.
El compromiso a nivel digestivo estuvo dado por la presencia de enterorragia y de una enteropatía perdedora de proteínas que llevó a una hipoproteinemia severa con anasarca. La enteropatía perdedora de proteínas se caracteriza por una pérdida de proteínas plasmáticas por el tubo digestivo que se confirmó por la dosificación de alfa-1-antitripsina en materias fecales que mostró valores elevados. Se descartaron las pérdidas extradigestivas. La enteropatía perdedora de proteínas se produce en este caso por aumento de la permeabilidad vascular. Se considera que el daño del epitelio vascular se produce por el depósito de inmunocomplejos a dicho nivel que activan el complemento, originándose sustancias quimiotácticas capaces de atraer células inflamatorias que lesionan la pared vascular provocando el pasaje de proteínas a la luz intestinal e hipoproteinemia (13). En otros pacientes los mecanismos del anasarca en la DMJ no son claros. En los casos publicados hasta la fecha que son 19 (14,15), la instalación de anasarca coincide con la progresión de la debilidad muscular y la disminución de las masas musculares sugiriendo un daño del endotelio capilar a nivel muscular provocado por el mismo mecanismo descrito con trasudado plasmático al intersticio. Es una manifestación rara, pero cuando existe se asocia a severa actividad de la enfermedad (14,15).
Desde el punto hematológico se destaca la presencia de plaquetopenia de mecanismo inmunológico (1).
Se descartó en este paciente la presencia de calcinosis mediante la clínica y realización de radiografías de partes blandas. Éstas aparecen en un 50% de los pacientes, surgiendo desde los seis meses de evolución de la enfermedad o en forma más tardía, tras diez a veinte años del inicio de la afección. La calcinosis consiste en la aparición de calcificaciones en la piel, el tejido subcutáneo, la grasa y las fascias intermusculares. Aparecen en forma de placas subcutáneas o nódulos en músculos y articulaciones. Estos depósitos tópicos de calcio contienen proteínas ricas en 4-carboxi-L-acetil-glutámico, un aminoácido transportador de calcio que se genera en exceso en estos pacientes. Las calcificaciones una vez que surgen pueden mostrar resolución espontánea o drenar en forma de exudado caseoso blanco y dejar cicatrices. Suelen presentarse en los casos de curso grave y se asocian con una alta incidencia de infecciones estafilocócicas por la alteración a nivel de la quimiotaxis de los granulocitos (1,8,11).
En cuanto a la terapéutica destacamos que se basa en inmunosupresores, soporte nutricional y fisioterapia. Los corticoides son los fármacos de elección (1,8) ya que han descendido notoriamente la morbimortalidad de estos pacientes. En este paciente no se comenzó con los mismos, dada la serología positiva para citomegalovirus y hasta obtener el resultado negativo del PCR para dicho agente. Fue necesaria la utilización de bolos de metilprednisolona así como de citostáticos como la ciclofosfamida por la refractariedad inicial al tratamiento, con grave cuadro respiratorio. Se utilizó también gammaglobulina intravenosa que ha demostrado eficacia en el manejo de estos enfermos. Una vez que el paciente mostró mejoría clínica y enzimática se comenzó a administrar prednisona más metrotexate; esta asociación ha permitido ir disminuyendo progresivamente la dosis de corticoides y los efectos adversos de los mismos. El objetivo es mantener la menor dosis de corticoides que permita que la DM persista en remisión (1).
En cuanto al pronóstico de la dermatomiositis juvenil ha mejorado de forma ostensible desde el comienzo del uso de los corticoides en el tratamiento. Este paciente presentaba un pronóstico muy grave al inicio del cuadro por la severa afectación respiratoria y digestiva. La inmunosupresión enérgica con corticoides y ciclofosfamida, así como el uso de altas dosis de gammaglobulina intravenosa lograron una repuesta favorable.
Como medidas adicionales deben tenerse en cuenta la fotoprotección y el uso de antimaláricos como la hidroxicloroquina, útil sobre todo en las manifestaciones cutáneas y articulares.
Se han descrito varias formas evolutivas en el curso clínico de la enfermedad:
a) Aguda o monocíclica: descrita en el 20% de los casos (1), presenta afectación limitada, buena respuesta a los esteroides y recuperación total en un plazo de 8 meses a 2 años (1,8).
b) Crónica ulcerativa o continuada: en el que la enfermedad se mantiene activa durante más de dos años, con ulceraciones cutáneas y gastrointestinales; calcinosis. Presenta exacerbaciones a pesar de mantener tratamiento continuado con corticoides (1,8).
c) Crónica no ulcerativa o policíclica: caracterizada por la aparición de recaídas después de retirar el tratamiento. Responden bien al tratamiento corticoideo inicial y toleran la disminución de la dosis y su suspensión. Posteriormente, tras un período de remisión variable (meses a varios años) aparece una nueva recaída (1,8).
Este paciente podrá presentar tanto una evolución monocíclica como crónica policíclica. Los controles de enzimas musculares periódicos son importantes ya que su elevación puede preceder un empuje clínico de la enfermedad.
La mortalidad de la DMJ es de 5 a 10%, ocurriendo sobre todo en la fase aguda de la enfermedad. Ésta se ocasiona por debilidad muscular progresiva, por refractariedad al tratamiento que ocasiona insuficiencia respiratoria; perforación gastrointestinal secundaria a ulceraciones ocasionada por vascultitis, miocarditis, sepsis y neumonía.
Las complicaciones infecciosas son causa frecuente de morbimortalidad en estos pacientes inmunodeprimidos por la enfermedad y su tratamiento.
Citas bibliográficas
1. Pascual MP, Montes de Oca F. Dermatomiositis juvenil. Acta Pediatr Esp 2002; 60 (10): 583-95.
2. Feber I. Conectivopatías: Dermatomiositis. En: Moraga F, ed. Protocolos de dermatología. [En línea] Madrid: Asociación española de Pediatría, 2003 < http://www.aeped.es/protocolos/dermatología > [consulta 15/abr/2005].
3. Martín Mateos MA, Cruz M. Dermatomiositis. En: Cruz M. Tratado de Pediatría. 8 ed. Madrid: Ergon, 2001: 348–9.
4. Pachaan LM. Dermatomiositis juvenil. En: Behrman RE, Kli RM, Hal B. Jeanson HB. Nelson. Tratado de Pediatría. 17 ed. Madrid: Elsevier, 2004: 813-6.
5. Fabius S, García de Larre-Borges A, Flain M, Blanco T, Chiosoni M. Dermatomiositis. Arch Pediatr Urug 1969; 7: 164-9.
6. Silver RM, Maricq HR. Estudios microvasculares seriados en la dermartomiositis infantil. Pediatrics (ed. esp) 1989; 27(2): 99-103.
7. Lauren M, Cooke P, Cooke N. Juvenile dermatomyositis: a clinical and inmunologic study. J Pediatr 1980; 96(2): 226-34.
8. Cassidy J, Petty R. Juvenile Dermatomiositis. En: Pediatric Rheumatology. 3 ed. Philadelphia: Saunders, 1995: 323-58.
9. Park S, Nyhan WL. Fatal pulmonary involvement in dermatomyositis. Am J Dis Child 1975; 129: 723-6.
10. Hoffman AH, Bernreuter W, Alarcón GS. Juvenile Dermatomiositis in a 28 month old Infant. Clin Pediatr 2003; 42: 375-8.
11. Sell-Salazar F. Dermatomiositis juvenil. Rev Neurol 2002; 34 (2):178- 80.
12. Kobayashi I, Ono S, Kawamura N, Okano M, Miyazawa K, Shibuya H, et al. IL-6 is a potential marker for interstitial lung disease associated with juvenile dermatomyositis. J Pediatr 2001; 138 (2): 274-6.
13. Camarero Salcés C. Enteropatía pierde proteínas. En: Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica. Tratamiento en Gastroenterología, Hepatología y Nutrición Pediátrica. Madrid: Ergon, 2004: 171-6.
14. Jeanne P, Mitchell GJ, Gregory J, Rider D, Rider L. Juvenile dermatomyositis presenting with anasarca: a possible indicator of severe disease activity. J Pediatr 2003; 138: 942-45.
15. Mehendiratta S, Banerjee P. Juvenile Dermatomyositis presenting with anasarca. Indian Pediatr 2004; 41:752- 53. X.
Correspondencia: E-mail: drsolepandolfo@hotmail.com
