










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.76 no.3 Montevideo 2005
ARTÍCULO ORIGINAL
Arch Pediatr Urug 2005; 76(3)
Colestasis neonatal: evolución de los pacientes diagnosticados entre 1983 y 2000 en dos servicios gastroenterológicos de referencia de Montevideo
DRES. VIRGINIA MéNDEZ 1, MARINA BURASTERO 2, MARíA NOEL CUADRO 2, LUCíA CASAMAYOU 3, CARMEN GUTIéRREZ 4, ALICIA MONTANO 5, GRACIELA CABALLERO 6, VIOLETA SERENO 7, HéCTOR PACHECO 8, RUVER BERAZATEGUI 9, OSCAR CHAVARRíA 10
1. Prof. Agda. de Clínica Pediátrica. Gastroenteróloga.2. Pediatra. Ex Residente de Pediatría.
3. Ex Residente de Pediatría.
4. Patóloga pediátrica. Jefe del Servicio de Anatomía Patológica Pediátrica del Ministerio de Salud Pública.
5. Prof. de Clínica Pediátrica.
6. Pediatra. Gastroenteróloga.
7. Pediatra.
8. Prof. Agdo. de Cirugía Pediátrica
9. Prof. de Cirugía Pediátrica.
10. Ex Prof. de Cirugía Pediátrica.
Servicio de Gastroenterología, Departamento de Especialidades Médico Quirúrgicas (DEMEQUI), Banco de Previsión Social (BPS), Servicio de Gastroenterología, Clínica Pediátrica A, Centro Hospitalario Pereira Rossell (CHPR). Montevideo. Uruguay
Fecha recibido: 2 de setiembre de 2005.
Fecha aprobado: 5 de diciembre de 2005.
Resumen
Con el objetivo de conocer la evolución de los pacientes con colestasis neonatal en relación a las diferentes etiologías y compararla con la de otros centros, se realizó un estudio descriptivo de los 51 niños con colestasis neonatal diagnosticados en los Servicios de Gastroenterología del Departamento de Especialidades Médico Quirúrgicas (DEMEQUI) y del Centro Hospitalario Pereira Rossell (CHPR) en el período comprendido entre 1983 y 2000 inclusive. Los resultados pretenden ser un aporte a la planificación de acciones futuras que mejoren la asistencia de estos niños.
Los datos se extrajeron de fichas precodificadas que se habían completado al asistir a los pacientes por primera vez y de las historias clínicas. En el 2001 los niños fueron citados para una entrevista en la que se realizó anamnesis, examen físico y estudios paraclínicos, registrando los datos en fichas codificadas que se analizaron con Epi Info 6.
Se analizaron y describieron las etiologías encontradas así como la evolución clínica y paraclínica. La evolución de los pacientes fue similar a la descrita enotras series excepto en el caso de los niños transplantados que tuvieron peor evolución y en los casos de hepatitis neonatal que presentaron todos buena evolución, probablemente porque varios fueron hepatitis neonatales transitorias.
Se destaca la derivación tardía al tercer nivel de atención. Se propone trabajar este tema en desarrollo profesional médico continuo, lograr mejor seguimiento y apoyo nutricional de los pacientes y conseguir los recursos para organizar un equipo de especialistas pediátricos que asuma el seguimiento de estos niños en el país, en forma centralizada, trabajando en coordinación con el equipo del centro de trasplante del exterior, mientras esta técnica no se realice en el país.
Palabras clave: COLESTASIS
ATRESIA BILIAR
RECIEN NACIDO
Summary
This is a descriptive study conducted to determine the course of patients with a history of neonatal cholestasis (NC) with regard various conditions, and to compare it with children’s course at other centres. The children enrolled had been diagnosed NC at the Gastroenterology Department of DEMEQUI and CHPR in the 1983-2000 period.
Data were drawn from pre-coded cards completed when patients were seen for the first time and from their clinical records. The children were summoned for an appointment where their clinical case, physical examination and lab testing were recorded in coded cards. Data were analyzed with Epi Info 6.
NC was diagnosed in 51 children from 1983 to 2000; 37 in DEMEQUI and 14 in CHPR. The etiologies, clinical and laboratory testing are discussed and described. Patients’ course was similar to that described in other series, except for transplanted children, who had a worse course, and the cases of neonatal hepatitis, all of which had a good course, probably because several had transient neonatal hepatitis.
Referral to tertiary care was typically late and should be especially addressed at continuing medical education activities. There is a need to promote a better follow-up and nutritional support for patients and to obtain the resources required to organize a team of paediatric experts to monitor these children in the country before and after transplantation, centralizing work and coordinating with the transplantation centre team abroad, until this technique is covered in Uruguay
Key words: CHOLESTASIS
BILIARY ATRESIA
INFANT, NEWBORN
Introducción
El diagnóstico y el tratamiento de la colestasis neonatal constituyen un gran desafío para el pediatra, dado que la colestasis puede ser la manifestación inicial de un grupo de enfermedades muy heterogéneo como alteraciones anatómicas intra o extrahepáticas, errores congénitos del metabolismo, infecciones y otras etiologías.
La posibilidad de realizar un tratamiento específico médico o quirúrgico hace, en algunos casos, necesario y urgente identificar la causa de la colestasis, aunque eso no siempre es posible (1).
La atresia de la vía biliar y la hepatitis neonatal idiopática, constituyen del 55 al 65% de todos los casos de colestasis neonatal (2). Estas dos entidades deben ser diferenciadas rápidamente, ya que su manejo terapéutico, evolución y pronóstico difieren significativamente (1).
La atresia de la vía biliar tiene una incidencia de un caso cada 8.000 a 15.000 recién nacidos vivos. Cuando se ha llegado al diagnóstico de atresia de la vía biliar se debe realizar rápidamente el tratamiento quirúrgico. En 75 a 85% de los casos la lesión encontrada es la obstrucción ductal en el hilio hepático o por arriba de él (tipo III) formando un cono de tejido fibroso denso que cubre todo el área perihiliar; lesión que se puede tratar con la hepaticoportoenterostomía de Kasai (1). Esta intervención es efectiva a largo plazo (5 años) en cerca de 40 a 50% de los casos, considerando los pacientes en los que se realiza como único procedimiento, o como una solución temporaria hasta el trasplante (3-5). La evolución postoperatoria de estos pacientes puede ser muy variable, dependiendo de varios factores, siendo la edad al momento de la cirugía un factor clave, obteniéndose flujo biliar en 80% de los casos si se opera antes de los 60 días de vida; la tasa de éxito decrece a 20% cuando la operación se realiza después de los 90 días de vida, según la mayoría de los autores (1,2,6). El restablecimiento del flujo biliar no equivale a curación. Las complicaciones postoperatorias son muy frecuentes. La colangitis ascendente se presenta en 40 a 60% de los pacientes con reparación exitosa, disminuyendo su frecuencia entre el primer y segundo año postoperatorio (1,3-5,7-9). La hipertensión portal se presenta en aproximadamente 70% de los pacientes y comienza a aparecer generalmente entre el segundo y tercer año postoperatorio (3); siendo la manifestación más frecuente el sangrado digestivo por várices esofágicas (5,8).
Los pacientes que evolucionan bien con la operación de Kasai y que no requieren trasplante hepático, constituyen solamente un 20 a 30% (3,5,6).
La sobrevida de los pacientes a los que no se les realiza operación de Kasai es menor a 2 años (sin trasplante) (2,10).
Las indicaciones para el trasplante hepático en esta patología son: Kasai no exitoso (sin pasaje de bilis al intestino); pacientes en que no se realizó Kasai por diagnóstico tardío; hepatopatía progresiva grave a pesar de operación de Kasai inicialmente exitosa; colangitis reiteradas, y falla de crecimiento (11).
En cuanto a la hepatitis neonatal, se distinguen dos tipos: hepatitis neonatal idiopática esporádica y la familiar, que representan el 80 y 20% de los casos respectivamente (2).
La evolución de los casos de hepatitis neonatal idiopática es muy variable. Los factores que favorecen la perpetuación del proceso colestásico y de la injuria hepatocítica no se conocen completamente aún.
De los casos esporádicos publicados por Danks y colaboradores, aproximadamente 60% se recupera, 10% evoluciona a enfermedad hepática crónica y 30% muere (12). Deutsch y colaboradores (13) obtuvieron resultados similares, pero otras series como las de Odievre y colaboradores (14) y Chang y colaboradores (15) tuvieron mejor pronóstico, con recuperación del 85%, un 5% desarrolla enfermedad hepática crónica y 10% muere.
La evolución es muy diferente en los casos de hepatitis neonatal familiar y su pobre pronóstico podría estar relacionado con la presencia de alteraciones congénitas o metabólicas subyacentes. En este grupo se describen en la actualidad las colestasis intrahepáticas familiares progresivas (PFIC), que incluyen la colestasis antes conocida como síndrome de Byler, clasificándoselas hoy en cuatro tipos y en las cuales se ha determinado el origen genético (2).
Otra entidad que podría discriminarse actualmente de los casos de hepatitis neonatal idiopática es la colestasis neonatal transitoria, que según Jacquemin constituyen el 8% de los casos de colestasis. La misma resulta de la asociación de inmadurez de la secreción biliar por prematurez y agresiones perinatales que llevan a hipoxia e isquemia hepática. Tiene resolución clínica espontánea en 3-5 meses y normalización de los parámetros bioquímicos en 10 meses (16).
Se describen cada vez más etiologías específicas en niños con colestasis neonatal, como los defectos en el metabolismo de los ácidos biliares que están actualmente bien identificados y que por lo tanto dejan de catalogarse como hepatitis neonatal idiopática (2).
Los pacientes con escasez sindrómica de conductos biliares o síndrome de Alagille tienen un curso clínico caracterizado por episodios recurrentes de colestasis. En un estudio de 80 casos, 28 (35%) tenían colestasis severa y permanente durante los cuatro primeros años de vida; disminuyendo la colestasis clínica a partir de los cinco años, con persistencia sólo de signos bioquímicos de colestasis y tasa de sobrevida de 85% (17). En la serie de Hoffenberg no se comprobó un pronóstico tan favorable, 50% de los pacientes requirieron trasplante hepático o murieron antes de los 19 años (18). A corto plazo la principal causa de mortalidad es la cardiopatía congénita compleja y a largo plazo la principal causa de morbilidad y mortalidad son las complicaciones hepáticas (2).
La escasez de conductos no sindrómica es un desorden muy raro de etiología desconocida, pero que podría tratarse de una “forma frustra” del síndrome de Alagille, con marcada fibrosis y rápida progresión hacia la cirrosis biliar y el fallo hepático (19).
Con respecto al déficit de á1-antitripsina, Psacharopoulos y colaboradores encontraron que a los 17 años un tercio de los pacientes había muerto, otro tercio había desarrollado cirrosis y sólo el 20% tenía recuperación clínica y bioquímica documentada (20). Nebbia y colaboradores demostraron que el curso de la enfermedad hepática es altamente variable, con evolución bimodal; la mitad desarrollan cirrosis y la otra mitad tiene evolución favorable a largo plazo (21).
El trasplante hepático en pediatría es, en la actualidad, una terapéutica estandarizada para el tratamiento de la insuficiencia hepática aguda y crónica. Más del 50% de los trasplantes en la edad pediátrica se deben a enfermedades colestásicas y dentro de éstas la causa más frecuente es la atresia de la vía biliar, seguida por las enfermedades metabólicas con predominio del déficit de á1-antitripsina. Desde 1980 ha habido una franca mejoría en los resultados de los trasplantes hepáticos pediátricos, en 1997 la sobrevida a 5 años alcanzaba el 80% de los casos en todos los centros de referencia. El éxito terapéutico se debe al mejor manejo de la medicación inmunosupresora, al avance de las técnicas quirúrgicas y al desarrollo de los centros de cuidado intensivo pediátricos (22,23).
Objetivo
Analizar la evolución de los pacientes con colestasis neonatal, que fueron diagnosticados y/o se controlaron en los Servicios de Gastroenterología del Centro de Especialidades Médico-Quirúrgicas (DEMEQUI) del Banco de Previsión Social (BPS) y de la Clínica Pediátrica “A” del Centro Hospitalario Pereira Rossell (CHPR) entre el año 1983 y 2000 y compararla con la de centros internacionales. Los resultados obtenidos pretenden ser un aporte a la planificación de acciones futuras que ambos centros podrán realizar para mejorar la asistencia que brindan a estos niños.
Material y método
Se realizó un estudio descriptivo de los pacientes con colestasis neonatal que se controlaron en los Servicios de Gastroenterología del BPS y de la Clínica Pediátrica “A” del CHPR durante el período comprendido entre 1983 y 2000 inclusive.
Se consideró que presentaban colestasis neonatal los niños que tenían en el primer trimestre de la vida, hiperbilirrubinemia con fracción conjugada mayor al 20% del total.
Los datos se extrajeron de las fichas de colestasis precodificadas que se habían completado al atender al paciente por primera vez, así como de las historias clínicas y se citó a todos los pacientes en el año 2001 para una entrevista personal realizándoles hepatograma, proteinograma electroforético, tiempo de protrombina y hemograma.
Los datos se registraron en una ficha diseñada a tal efecto, que consta de dos partes, una primera donde se incluyeron: ficha patronímica; antecedentes perinatales; manifestaciones clínicas; diagnóstico etiológico; tratamiento realizado; evolución clínica; paraclínica y complicaciones, y una segunda parte que se completó durante la entrevista personal, en donde se consignó el estado actual del paciente (se consideraron como actual los últimos 6 meses): presencia de síntomas y signos; desarrollo neurológico; examen físico y paraclínica actual. Se definió buen estado nutricional cuando el score Z de peso, talla y relación peso/talla se encontraba entre ± 2 y la velocidad de crecimiento era normal.
El diagnóstico anatomopatológico de atresia de vía biliar se realizó por el hallazgo histológico característico: alteración portal con aumento del número de los conductos biliares interlobulares e intensa proliferación ductular con colestasis ductular; afectación de todos los espacios porta con expansión del tejido fibroso portal y puentes fibrosos que unen los espacios portales (28-30). El diagnóstico se confirmó por laparotomía y por antomía patológica de la biopsia obtenida durante la cirugía en todos los pacientes a los que se realizó operación de Kasai y en el hígado resecado en los niños en los que se hizo transplante hepático.
La hepatitis neonatal idiopática se definió por los cambios histológicos característicos a nivel lobulillar: edema y vacuolización hepatocítica, transformación gigantocelular, colestasis hepatocítica y canalicular, así como focos de hematopoyesis extramedular, sin colestasis ductal y ductular y sin fibrosis lobulillar y portal (31-33) en un niño sin etiología demostrable de su hepatitis.
Para el diagnóstico de síndrome de Alagille se siguieron los criterios mayores de Alagille: 1) escasez de conductos biliares en una biopsia hepática (índice de conductos biliares/espacios porta menor de 0,5); 2) facies característica; 3) malformaciones cardíacas; 4) anomalías óseas sobre todo hemivértebras y vértebras en ala de mariposa, y 5) embriotoxón posterior (17,18). Se diagnosticó este síndrome con la presencia de al menos tres de los cinco criterios mayores.
El diagnóstico de escasez no sindrómica de conductos biliares se realizó cuando había escasez de conductos biliares en la biopsia hepática (índice de conductos biliares/espacios porta menor de 0,5), no cumplía con tener al menos tres criterios de Alagille y se descartaron las otras causas de colestasis (19).
El déficit de á1-antitripsina se consideró cuando la dosificación de á1-antitripsina por técnica de inmunodifusión radial fue menor a 200 mg/dl, y en la anatomía patológica de la biopsia hepática se objetivó la presencia de glóbulos PAS+, diastasa resistentes en el citoplasma de los hepatocitos del limite portolobulillar, si bien este hallazgo no es patognomónico (20,21).
La galactosemia se diagnosticó por dosificación de galactosa 1-fosfato uridil treansferasa (en glóbulos rojos) (20,21).
El diagnóstico de colestasis por alimentación parenteral se basó en el hallazgo histológico de esteatosis, estasis biliar predominante en la región centrolobular y en las células de Kupffer, en los pacientes sometidos a más de dos semanas de alimentación parenteral total y en los que se descartaron otras causas de colestasis.
La hepatitis por citomegalovirus se diagnosticó por serología positiva (IgM) para citomegalovirus, ecografía transfontanelar con macrocalcificaciones en los núcleos de la base, fondo de ojo con hemorragias retinianas y disfunción del VIII par en potenciales evocados (2).
El quiste de colédoco se diagnosticó por ecografía (2) y se confirmó en el acto quirúrgico.
Los transplantes realizados antes de 1993 se hicieron en distintos centros, luego de este año debido al convenio que el Fondo Nacional de Recursos de Uruguay suscribió con el Hospital Italiano de Buenos Aires, estos se realizaron allí, excepto uno que se hizo en Brasil. El seguimiento postransplante se realizó en el Hospital Italiano de Buenos Aires en los niños transplantados luego de 1993 y en el CHPR y en DEMEQUI respectivamente en dos niños transplantados antes de 1993.
Los datos se analizaron con el programa Epi Info 6.
Resultados
En el período comprendido entre 1983 y 2000 inclusive, se diagnosticó colestasis neonatal en 51 pacientes, 27 de sexo femenino. Treinta y siete de ellos consultaron en BPS y 14 en el CHPR. Del total, 10 pacientes fallecieron y de los 41 que están vivos, fueron entrevistados 31.
La mediana de edad de los pacientes vivos fue al momento de la entrevista, de 5 años y 1 mes, con un rango de 8 meses a 18 años y 10 meses.
La etiología de la colestasis se muestra en la tabla 1.
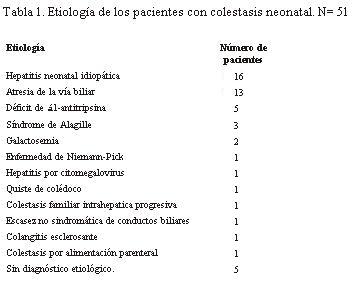
Los 10 pacientes fallecidos fueron: cinco portadores de atresia de via biliar, uno con síndrome de Nieman-Pick, uno con de quiste de colédoco, dos con síndrome de Alagille, uno con colestasis familiar progresiva.
Atresia de vía biliar
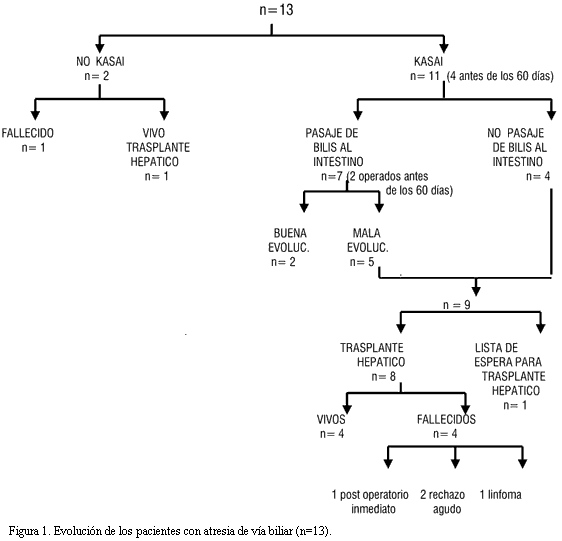
La evolución de los pacientes con atresia de la vía biliar se muestra en la figura 1.
De los 13 pacientes con atresia de la vía biliar, siete eran de sexo femenino y seis de sexo masculino.
Se encontró asociación con malformaciones congénitas en un paciente, que presentaba una malrrotación intestinal, correspondiendo entonces a la forma embrionaria de atresia de la vía biliar.
Dos de los 13 pacientes no fueron operados con técnica de Kasai. En una paciente porque la biopsia sugirió en dos oportunidades (una de ellas por laparoscopía) hepatitis neonatal y no se obtuvo autorización para realizar exploración quirúrgica ni reiteración de la biopsia cuando se sospechó clínicamente atresia de vía biliar. Evolucionó a hipertensión portal e insuficiencia hepatocítica y requirió trasplante a los siete meses de vida.
Al otro paciente no se le realizó operación de Kasai, ya que en la laparotomía exploradora no se encontró placa hiliar y no pudo realizarse ningún tipo de derivación, falleció a los 13 meses de vida sin ser trasplantado.
A los 11 pacientes restantes se les realizó operación de Kasai, presentando una mediana de edad en el momento de la intervención quirúrgica de 65 días (rango 51-90 días). Se destaca que sólo en cuatro pacientes se realizó la intervención antes de los 60 días de vida.
La operación de Kasai fue exitosa, es decir que se obtuvo pasaje de bilis al intestino, en siete de los 11 pacientes. En los cuatro restantes, no se obtuvo pasaje de bilis al intestino (tabla 2).

De los siete pacientes con pasaje de bilis al intestino, dos evolucionaron bien solo con el Kasai. Los cinco restantes, al igual que los cuatro en los que la operación no fue exitosa (en total nueve pacientes), desarrollaron signos y síntomas de hipertensión portal con ascitis (n=6), sangrado de várices esofágicas (n=3), síndrome hepato-pulmonar (n=4), desnutrición (n=7). De estos 9 pacientes, 8 fueron trasplantados y uno, de 10 meses de edad, quedó en lista de espera. La mediana de edad al momento del trasplante fue de 1 año y 8 meses (rango 1 año y 8 días a 3 años y 6 meses). El estado nutricional previo al trasplante fue adecuado sólo en dos pacientes, el resto estaban desnutridos, en algunos casos en forma muy severa (tabla 3).
De los ocho pacientes trasplantados, fallecieron cuatro: uno en el postoperatorio inmediato, dos por rechazo agudo, uno por linfoma retroperitoneal (a los cuatro años del trasplante habiendo tenido una buena evolución postrasplante hasta la aparición del tumor).
Los cuatro pacientes restantes que viven presentaron mejoría del estado nutricional a los tres meses del trasplante (mediana) y la paraclínica se normalizó al mes y 15 días (mediana).
De los 11 pacientes con operación de Kasai ocho tuvieron episodios de colangitis, algunos de ellos se confirmaron con anatomía patológica. Con el tratamiento antibiótico instituido tuvieron remisión clínica y paraclínica.
Como ya se ha expresado dos de los 11 pacientes que fueron intervenidos con la técnica de Kasai, evolucionaron bien y no requirieron trasplante Se obtuvo pasaje de bilis al intestino desde el postoperatorio inmediato, la ictericia, coluria y acolia desaparecieron aproximadamente a los dos meses de la operación, la paraclínica se normalizó alrededor de los seis meses del postoperatorio, han presentado como complicación episodios de colangitis (tres en un niño y uno en el otro), que remitieron con tratamiento antibiótico.
Estado actual de los pacientes
De los 13 pacientes con diagnóstico de atresia de vía biliar, ocho se encuentran vivos; dos que han tenido buena evolución sólo con la técnica de Kasai, cinco que han sido trasplantados (cuatro con Kasai y otro sin la intervención) y uno se encuentra en lista de espera para ser trasplantado.
Los dos pacientes que fueron tratados exclusivamente con Kasai tienen 1 año 4 meses y 5 años 3 meses respectivamente. Ambos presentan buen estado nutricional. Uno de ellos tiene hepatomegalia leve y alteraciones bioquímicas leves de colestasis y el otro está asintomático y sin alteraciones bioquímicas.
Los cuatro pacientes con intervención de Kasai que fueron trasplantados, tienen: 4 años 3 meses (9 meses de trasplantado); 3 años 10 meses (8 meses de trasplantado); 3 años (2 años de trasplantado), y 1 año 2 meses (2 meses de trasplantado). Actualmente tienen buen estado nutricional y clínica y paraclínica normal.
La paciente que fue trasplantada a los siete meses de edad, sin haberse realizado la hepaticoportoenterostomía de Kasai, tiene actualmente 3 años 3 meses. Presenta buen estado nutricional, clínica y paraclínica normal. Se destaca que esta paciente proviene de un medio socio-económico muy deficitario que ha determinado que no se cumpla el tratamiento en forma adecuada y que padezca enfermedades sociales como episodios de diarrea aguda infantil, así como caries dentales múltiples.
Hepatitis neonatal idiopática
De los 16 pacientes con hepatitis neonatal idiopática, 10 presentaban antecedentes perinatales de sufrimiento fetal agudo con depresión neonatal severa (n=8), prematurez (8), sepsis (n=4), hipoglicemias (n=2), enfermedad hemolítica por conflicto Rh (n=2).
La mediana de duración de la ictericia fue de dos meses (rango 1 mes - 10 meses). En los ocho pacientes que tuvieron exámenes paraclínicos seriados, la paraclínica se había normalizado entre los tres y cuatro meses de edad. Ninguno presentó complicaciones.
Se entrevistaron 12 pacientes de los 16, con una mediana de edad de 4 años 1 mes (rango 11 meses a 11 años 7 meses). Todos se encuentran asintomáticos y con paraclínica normal. Diez tienen buen estado nutricional y dos pacientes, con encefalopatía crónica, presentan desnutrición crónica compensada.
Síndrome de Alagille
De los tres pacientes con síndrome de Alagille fallecieron dos. Uno de los pacientes falleció a los 10 meses de edad a causa de una descompensación de su cardiopatía congénita (atresia pulmonar y comunicación interventricular, que había requerido intervención quirúrgica paliativa con la técnica de Blalock a los 13 días de vida).
El otro paciente falleció a los 21 meses de edad. Había desarrollado insuficiencia hepatocítica e hipertensión portal severa y falleció por hemorragia pulmonar previo a la realización del trasplante.
El paciente que se encuentra vivo tiene cuatro años y buen estado nutricional. Clínicamente presenta prurito leve, signo de papel moneda, palmas hepáticas y hepatomegalia de consistencia aumentada. En la paraclínica se destaca aumento de la bilirrubina directa y de la fosfatasa alcalina. Está siendo tratado desde los 2 años y 6 meses con ácido ursodesoxicólico, obteniéndose mejoría parcial del prurito.
Deficit de a1-antitripsina
Se diagnosticó déficit de a1-antitripsina en cinco pacientes que están todos vivos (tabla 4).
Tres de los pacientes desarrollaron signos clínicos de hipertensión portal. Uno de ellos comenzó con los primeros signos de hipertensión portal a los dos años de edad, agregó hiperesplenismo a los 8 años, permaneciendo con función hepática normal hasta los 13 años. Actualmente tiene 14 años, presenta desnutrición, esplenomegalia y várices esofágicas grado II que no han sangrado, con elementos de insuficiencia hepatocítica. Tiene indicación de trasplante.El segundo paciente tiene 18 años, comenzó con hipertensión portal a los 6 años de edad, agregó hiperesplenismo a los 12 años. Presenta actualmente esplenomegalia de 7 cm del reborde costal de consistencia aumentada, sin elementos de insuficiencia hepatocítica y excelente estado nutricional. El tercer paciente también de 18 años, comenzó con signos de hipertensión portal a los 4 meses, presentó sangrado por várices esofágicas en varias oportunidades (la primera vez a los 17 meses). En la evolución instaló insuficiencia hepatocítica, por lo cual fue trasplantado a los 5 años y 6 meses. Actualmente tiene buen estado nutricional, se encuentra clínicamente normal. Esta recibiendo terapia inmunosupresora (ciclosporina y prednisona).
En el caso de los otros dos pacientes, uno tiene 7 años, buen estado nutricional, clínicamente presenta solo hepatomegalia y la paraclínica es normal. El otro tiene actualmente 12 años, buen estado nutricional y ha permanecido totalmente asintomático y con paraclínica normal.
Errores congénitos del metabolismo
Tres de los pacientes presentaron colestasis por errores congénitos del metabolismo, uno con enfermedad de Niemann-Pick tipo C y dos con galactosemia.
El paciente con enfermedad de Niemann-Pick murió a los 8 meses de edad a causa de disturbios metabólicos, con diagnóstico presuntivo de enfermedad metabólica. El diagnóstico postmorten fue enfermedad de Niemann-Pick tipo C.
Los dos pacientes con galactosemia presentaron al inicio ictericia, hepatomegalia, depresión neuropsíquica, hipotonía y cataratas. Entre el mes y los dos meses de edad la mayoría de los síntomas y signos desaparecieron espontáneamente, excepto la hepatomegalia y las cataratas.
En uno de los pacientes se inició el tratamiento libre de galactosa a los dos meses de edad, con desaparición de las cataratas y la hepatomegalia. El segundo paciente comenzó el tratamiento a los 9 meses de edad, con desaparición de las cataratas y disminución de la hepatomegalia.
Actualmente tienen 7 y 8 años respectivamente y buen estado nutricional. Uno de ellos presenta hepatomegalia leve de consistencia normal y el otro se encuentra clínicamente normal. Los exámenes paraclínicos ambos son normales. Cumplen estrictamente la dieta libre de galactosa.
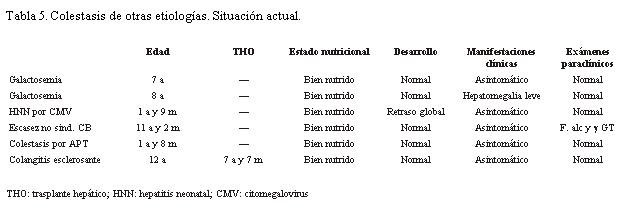
Otras etiologías
El paciente con hepatitis neonatal por citomegalovirus tiene 1 año y 9 meses y buen estado nutricional. Desde el punto de vista hepático evolucionó bien y la paraclínica es normal.
Presenta un retardo global del desarrollo
El paciente con colestasis por alimentación parenteral estuvo alimentado de esta forma desde el nacimiento hasta los 3 meses. Presentó acolia y coluria los 4 primeros meses y la ictericia persistió hasta los 7 meses. La paraclínica se normalizó en ese momento.
Actualmente tiene 1 año y 8 meses y buen estado nutricional. Se encuentra con clínica y paraclínica normal.
El paciente con escasez de conductos no sindrómica tuvo al inicio biopsia y clínica característica de hepatitis neonatal idiopática. Presentó ictericia hasta los 3 meses y luego permaneció asintomático hasta los 2 años. A esa edad comenzó con prurito, hepatomegalia y hepatograma alterado, por lo que se realizó nueva biopsia que mostró escasez de conductos biliares. Dado que no cumplía con los criterios de Alagille y se descartaron las otras causas de colestasis, se realizó el diagnosticó de escasez no sindrómica de conductos biliares.
Actualmente tiene 11 años y 2 meses, buen estado nutricional. Persiste con prurito que desaparece si recibe ácido ursodesoxicólico a 20 mg/kg El hepatograma siempre evidencia fosfatasa alcalina y ãGT elevadas.
El paciente con quiste de colédoco tipo I fue intervenido, realizándose anastomosis yeyuno-colédoco en Y de Roux. Presentaba hepatomegalia y esplenomegalia que fueron en aumento, ascitis y desnutrición severa. Falleció al año y 3 meses de vida por fallo respiratorio con diagnóstico necrópsico de neumonía.
El paciente con diagnóstico de colestasis intrahepática familiar progresiva de tipo II presentó ictericia, coluria, hipocolia y hepatomegalia desde el nacimiento. En la evolución la ictericia permaneció estacionaria, la coluria y la hipocolia persistieron en empujes y la hepatomegalia fue en aumento, desarrollando posteriormente esplenomegalia.
La biopsia a los tres meses de edad mostró patrón de hepatitis neonatal, la paraclínica para descartar otras causas de colestasis fue normal. Evolucionó rápidamente a la peoría, falleciendo al año y 2 meses a causa de insuficiencia hepática.
A la paciente con colangitis esclerosante se le realizó biopsia hepática a los 5 meses que mostró fibrosis hepática. A los 7 meses de edad se le realizo colecistohepaticostomía, persistiendo con ictericia y coluria. No consultó hasta los 4 años, momento que comenzó con sangrado masivo por várices esofágicas que se esclerosaron, presentando además signos de insuficiencia hepatocítica.
Se le realizó TAC de abdomen que mostró imagen quística en el parénquima hepático, lo que junto a la historia previa, hizo sospechar el diagnóstico de colangitis esclerosante. El mismo se confirmó por biopsia hepática.
Se transplantó a los 7 años y 7 meses con buena evolución postrasplante.
Tiene actualmente 12 años, buen estado nutricional y se encuentra con clínica y paraclínica normal (tabla 5).
Pacientes sin diagnóstico etiológico
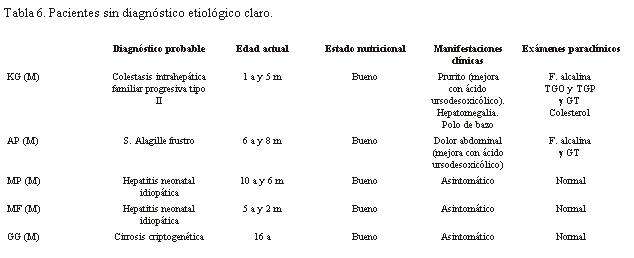
Hasta el momento hay cinco pacientes que no presentan un diagnóstico etiológico claro.
Uno de ellos podría tratarse de una colestasis intrahepática familiar progresiva.
Otro un síndrome de Alagille frustro, dado que presenta facies característica, la biopsia no fue concluyente (se repetirá, pues la escasez de conductos con frecuencia se hace evidente en la evolución) y no cumple con tener al menos tres de los criterios mayores diagnósticos del síndrome de Alagille. Otro de los pacientes mostró en la biopsia escasez de conductos biliares y luego tuvo buena evolución, por lo que podría corresponder a una hepatitis neonatal idiopática.
En un cuarto paciente por la biopsia se sospechó enfermedad metabólica, pero luego tuvo excelente evolución clínica y paraclínica, por lo cual se considera que pueda corresponder también a hepatitis neonatal idiopática.
El último paciente presentó una colestasis desde la etapa neonatal cuya etiología no fue aclarada, cuando concurrió a controlarse ya tenía una biopsia que mostraba cirrosis, la cual se catalogó entonces como criptogenética (tabla 6).
DiscusiónLos datos obtenidos de esta serie son a grandes rasgos similares a los de la bibliografía. La prevalencia relativa de las distintas patologías coincide con otros trabajos (1), la atresia de vía biliar y la hepatitis neonatal representan el 56% de la muestra.
Discutiremos algunos de los aspectos referidos a atresia de la vía biliar, hepatitis neonatal idiopática, déficit de á1-antitripsina y síndrome de Alagille.
No analizaremos las otras patologías que se describen en éste trabajo debido a que se presentaron en un número muy bajo de pacientes
Atresia de vía biliar
Los pacientes con atresia de vía biliar constituyen el 25% de todos los pacientes (1,2).
La relación masculino femenino fue de 1:1,2, planteándose en esta enfermedad un predominio en el sexo femenino.
Se describe asociación con otras malformaciones congénitas como poliesplenia, malrotación intestinal, malformaciones cardíacas y vasculares abdominales en un 20% de los casos (24), hecho que fue comprobado en uno de los pacientes que presentaba malrrotación intestinal.
Los hallazgos histológicos mostraron las alteraciones características de atresia de vía biliar en 11 pacientes, en los otros dos casos correspondieron al patrón de hepatitis neonatal en uno y escasez de conductos biliares en otro. La biopsia fue concordante con el diagnóstico en 85% de los casos como se menciona en otros trabajos (11).
A 11 de 13 pacientes se les realizó operación de Kasai. La mediana de edad en el momento de la cirugía fue de 65 días, similar a las de otras series revisadas (3,7,25). Solo cuatro pacientes se operaron, como se aconseja por la mayoría de los autores, antes de los 60 días de vida; lo que indica que no hay una referencia oportuna de estos niños.
Con respecto a las complicaciones en la evolución postoperatoria del Kasai, la colangitis ascendente se presentó en porcentaje similar a la serie de Lilly (3). La hipertensión portal también se presentó en número similar a lo descrito en otras series. Las várices esofágicas tuvieron una incidencia algo menor de la descrita, hecho que probablemente se debe a que no se realizó endoscopía a todos los pacientes y sólo se comprobaron las que se evidenciaron por sangrado. No se realizó endoscopía a todos los pacientes porque estaba pautado realizar la primera endoscopía a los 2 años de la operación de Kasai y la mayoría de los pacientes requirieron trasplante antes de esa edad. Debería evaluarse la conveniencia de adelantar el momento de la realización de la primera endoscopía, al año de realizada la intervención de Kasai.
El hiperesplenismo se presenta en 20% de los pacientes con Kasai (5,7,8,12,26), los pacientes analizados no lo presentaron y esto probablemente también se explica por el poco tiempo de evolución antes del transplante, lo que no permitió su desarrollo.
Los dos pacientes que evolucionan bien sólo con el Kasai tienen un seguimiento de 1 año y 6 meses y 5 años. Se han encontrado tasas de sobrevida sólo con el Kasai de 28% con un seguimiento de 1 a 16 años en la serie de Lilly y colaboradores, 30% con un seguimiento de 1 a 5 años en Yanchar y colaboradores, 23% con un seguimiento a 10 años en Karrer y colaboradores y 33% con un seguimiento a 10 años en Kasai y colaboradores.
En los pacientes analizados no se encontró la correlación descrita en muchos trabajos entre la edad tardía de la operación de Kasai y la no obtención de flujo biliar. Sin embargo, el grupo hepatológico del King´s College ha publicado, que los primeros 100 días de vida son los útiles para realizar la portoenterostomía de Kasai y no 60 días como refieren la mayoría de las publicaciones (4).
Los trasplantes hepáticos en los pacientes con atresia de vía biliar corregida con la intervención de Kasai se realizaron con una mediana de edad similar a la que plantean otros autores (4,23,27).
La mortalidad de los transplantados fue alta, de un 50%. Esto podría deberse a varias causas, en primer lugar por tratarse de una serie retrospectiva, algunos de los pacientes fallecidos habían sido trasplantados en los primeros años de la década del 90, cuando no existían algunas de las drogas inmunosupresoras que hay actualmente (5,15). Otra probable explicación es que la mayoría de los pacientes llegaron al trasplante con muy mal estado nutricional, en parte debido a pertenecer a un medio socio económico y cultural deficitario (que determina que presenten dificultades para concurrir a los controles y tengan carencias importantes de cuidados, vivienda y educación) y por otro lado los cuidados médicos no han sido lo suficientemente efectivos y estrictos como para lograr mantener un mejor estado nutriciónal en dichos pacientes. Por último los trasplantes se hicieron en diferentes centros y no hay un equipo profesional, en el país, que trabaje en forma protocolizada en el seguimiento pre y postrasplante de estos pacientes.
Parece imprescindible para mejorar la evolución de éstos pacientes, lograr recursos y centralizar el diagnóstico y tratamiento de estos niños en una unidad hepatológica pediátrica del tercer nivel de atención, para optimizar los cuidados pretrasplante y concretar el seguimiento postransplante en el país, en esta misma unidad.
Hepatitis neonatal idiopática
Con respecto a la evolución clínica de los pacientes que presentaron hepatitis neonatal idiopática, ninguno de ellos desarrolló enfermedad hepática crónica, ni murió, hecho que difiere con los trabajos ya referidos de Odievre (14) y Chang (15) que tenían 85% de recuperación, 10% de enfermedad hepática crónica y 5% de mortalidad y sobre todo con los de Danks ( 12) y Deutsch (13) con sólo 60% de recuperación, 10% de enfermedad hepática crónica y 30% de mortalidad. Si bien el número de pacientes es limitado, se piensa que esta diferencia pueda deberse a que en 10 de los pacientes, el diagnóstico que se realizó retrospectivamente es de hepatitis neonatal transitoria (entidad descripta por Jacquemin) (16). En esta última, la mediana de duración de la ictericia es de 3,5 meses, la paraclínica se normaliza a una mediana de edad de 10 meses y tienen generalmente buena evolución, datos que coinciden con la evolución de éstos pacientes.
Síndrome de Alagille
El número de pacientes es pequeño y el paciente que se encuentra vivo tiene recién cuatro años, por lo cual es muy difícil sacar conclusiones respecto a la evolución.
Al considerar los pacientes fallecidos, dos murieron en etapas tempranas de la enfermedad, uno a causa de su cardiopatía congénita, hecho que es coincidente con la bibliografía, y el otro a causa de complicaciones de hipertensión portal severa, hecho que no es frecuente en la evolución a corto plazo (sí lo es a largo plazo). La mala evolución que presentó este paciente dependió en gran medida del medio socio económico muy deficitario del que provenía, que determinó por un lado que no se controlara y cuidara adecuadamente y por otro que no pudiera realizarse el trasplante hepático en el momento adecuado.
Déficit de a1-antitripsina
Estos cinco pacientes tuvieron una evolución muy variable, como sucede habitualmente en esta enfermedad, aproximadamente la mitad evolucionaron a la cirrosis y un paciente se ha recuperado clínica y bioquímicamente. Estos resultados son similares a los de la serie de Nebbia (21) (tabla 4).
Trasplante hepático
Con respecto a los pacientes transplantados, la mayoría corresponden a atresia de vía biliar y en segundo lugar a déficit de á1-antitripsina, lo que concuerda con la bibliografía.
La mortalidad es mayor que la esperada, probablemente por el predominio de casos de atresia transplantados con alta mortalidad, hecho que ya fue discutido y que por diferentes motivos (medio del que provienen, cuidados médicos y nutricionales que no son óptimos, carencia de un centro único especializado y con recursos suficientes), estos pacientes llegan al transplante con mal estado nutricional y muchas veces más tarde de lo que sería deseable.
Tomando en cuenta estos resultados, se pudo visualizar claramente hacia dónde se deben canalizar los esfuerzos para mejorar la asistencia de los niños con colestasis neonatal:
1) Trabajar en el desarrollo profesional médico continuo, para que el pediatra y otros integrantes del equipo de salud conozcan más acerca de la colestasis neonatal y deriven precozmente a estos niños al tercer nivel de atención.
2) Lograr mejor seguimiento y apoyo nutricional de los pacientes, implementando programas de apoyo nutricional específicos para estos niños y mejorando la referencia y la contra referencia.
3) Conseguir los recursos necesarios y organizar un equipo de especialistas pediátricos para realizar el seguimiento pretransplante y luego del transplante en el Uruguay, en forma centralizada, en una unidad hepatológica pediátrica en el tercer nivel de atención, trabajando en coordinación con el equipo del centro de trasplante del exterior mientras esta técnica no se realice en Uruguay.
Bibliografía
1. Balistreri WF. Neonatal Cholestasis. Medical Progress. J Pediatr 1985; 106: 171-84.
2. Dellert S, Balistreri W. Neonatal cholestasis. In: Walker WA, Durie P, Hamilton J. Pediatric Gastrointestinal Disease. 3 ed. Ontario: Decker, 2000: 880-94.
3. Lilly J, Karrer F, Hall R , Stelling GP, Vazquez-Estevez JJ, Greenolz SK, et al. The surgery of biliary atresia. Ann Surg 1989; 210: 289-96.
4. Davenport M, Kerkar N, Mieli-Vergani G, Mowat AP, Howard ER. Biliary atresia: the King¨s College Hospital experience (1974- 1995). J Pediatr Surg 1997; 32: 479-85.
5. Karrer FM, Price MR, Bensard DD, Sokol RJ; Narkewicz MR, Smith DJ, et al. Long term results with the Kasai operation for biliary atresia. Arch Surg 1996; 131: 493-6.
6. Kasai M. Treatment of biliary atresia with special reference to hepatic portoenterostomy and its modifications. Prog Pediatr Surg 1974; 6: 5-52.
7. TaggeDU, Tagge EP, Drongowsky RA, Oldham KT, Coran AG. A long term experience with biliary atresia. Reassessment of prognostic factors. Ann Surg 1991; 214: 590-8.
8. Kobayashi A, Itabashi F, Ohbe Y. Long term prognosis in biliary atresia after hepatic portoenterostomy: analysis of 35 patients who survived beyond 5 years of age. J Pediatr 1984; 105: 243-6.
9. Battaglini G, Previtera C. Prognosi a lungo termine dei pazienti affetti da atresia delle vie biliari extrahepatiche operati con successo. Minerva Pediatr 1991; 43: 493-8.
10. Adelman S. Prognosis of uncorrected biliary atresia: an update. J Pediatr Surg. 1978; 13: 389.
11. Shah HA, Spivak W. Neonatal cholestasis. New approaches to diagnostic evaluation and therapy. Pediatr Clin North Am 1994; 4: 943-66.
12. DanksDM, Campbell PE, Smith AL. Prognosis of babies with neonatal hepatitis. Arch Dis Child 1977; 52: 368-72.
13. Deutsch J, Smith AL, Danks DM, Campbell PE. Long term prognosis for babies with neonatal liver disease. Arch Dis Child 1985; 60: 447–51.
14. Odrievre F, Hadchouel M, Landrieu P, Alagille D, Eliot N. Long term prognosis for infants with intrahepatic cholestasis and patent extrahepatic biliary tract. Arch Dis Child 1981; 56: 373-6.
15. Chang MH. Neonatal hepatitis:a follow up study. J Pediatr Gastroenterol Nutr 1987; 6: 203-7.
16. Jacquemin E, Lykavieris P, Chaoui N, Hadchouel M, Bernard O. Transient neonatal cholestasis: origin and outcome. J Pediatr 1998; 133: 563-7.
17. Alagille D, Estrada A, Hadchouel A, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr 1987; 110: 195-200.
18. Hoffenberg EJ, Narkewicz RM, Sondeheimer JM, Smith DJ, Silverman A, Sokol RJ. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr 1995; 127: 220-4.
19. Mowat AP. Liver disorders in childhood. 3 ed. Oxford : Butterworth Heinemann Eds, 1994.
20. Psacharopoulos HT, Mowat AP, Cook PJ, Carlile PA, Portmann B, Rodeck CH. Outcome of liver disease associated with alfa-1 antitrypsin deficiency (Pi Z): implications for genetic counseling and antenatal diagnosis. Arch Dis Child 1983; 58: 882-7.
21. Nebbia G, Hadchouel M, Odievre M, Alagille D. Early assessment of evolution of liver disease associated with alfa-1 antitrypsin deficiency in childhood. J Pediatr 1983; 102: 661-5.
22. Whitington PF, Balistreri WF. Liver transplantations in pediatrics: indications, contraindications, and pretransplant management. J Pediatr 1991; 118: 169-77.
23. Goss JA, Shackleton CR, Swenson MD, Satou NL, Nuesse BJ, Imagawa DK, et al. Orthotopic liver transplantation for congenital biliary atresia. An 11 year, single-center experience. Ann Surg 1996; 224: 276-87.
24. Yanchar NL, Shapiro AM, Sigalet DL. Is early response to portoenterostomy predictive of long- term outcome for patients with biliary atresia?. J Pediatr Surg 1996; 3: 774-8.
25. Altmann RP, Lilly JR, Greenfeld J, Weinberg A, van Leeuwen K, Flanigan L. A multivariable risk factor analysis of the portoenterostomy (Kasai) procedure for biliary atresia. Twenty-five years of experience from two centers. Ann. Surg 1997; 226: 348-55.
26. Ohi R, Nio M, Chiba T, Endo N, Goto M, Ibrahim M. Long term follow-up after surgery for patients with biliary atresia. J Pediatr Surg 1990; 25: 442-5.
27. Vacanti JP, Shamberger RC, Eraklis A, Lillehei CW. The therapy of biliary atresia combining the Kasai portoenterostomy with liver transplantation: a single center experience. J Pediatr Surg 1990; 25: 149-52.
28. Witzleben CL. Extrahepatic biliary atresia: Concepts of causes, diagnosis and management. Perspect Pediatr Pathol 1979; 5: 41-62.
29. Nietgen GW, Vacanti JP, Perez-Atayde AR. Intrahepatic bile duct loss in biliary atresia despite portoenterostomy: A consequence of ongoing obstruction? Gastroenterology 1992; 102: 2126-33.
30. Okamoto S, Oike F, Egawa H. Heterogeneity of the expanded liver at the time of transplantation after failed Kasai procedure for biliary atresia: an inmunohistochemical study of biliary system. Gastroenterology 1996; 24: 556A.
31. Montgomery CK, Rubner BH. Neonatal hepatocellular giant cell transformation: A review. Perspect Pediatr Pathol 1976; 3: 85-101.
32. Jevon GP, Dimmick JE. Histophatologic approach to metabolic liver disease: part I. Perspect Pediatr Pathol 1999; 21: 40-60.
33. Jevon GP, Dimmick JE. Histopatologic approach to metabolic liver disease: part II. Perspect Pediatr Pathol 1999; 21: 61-9.
34. Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental and sexual development, and cardiac murmur. J Pediatr 1975; 86: 63-71.
35. Witzelben CL. Bile duct paucity (“ Intrahepatic atresia”). Perspect Pediatr Pathol 1982; 7: 185-201.
Correspondencia: Dra. Virginia Méndez Amsterdam 1496. Montevideo 11400. Uruguay
E-mail: vimendez@adinet.com.uy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}