










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.75 no.4 Montevideo dic. 2004
CASO CLÍNICO
Arch Pediatr Urug 2004; 75(4): 323-326
Enfoque diagnóstico de la calcinosis cutánea.
A propósito de un caso
DRES. ANDREA SPREMOLLA 1, GLORIA MARINA SALMENTóN 2, VIRGINIA VALLS 3, JULIO BONASSE 4, GRISELDA DE ANDA 5
1. Médica pediatra.
2. Médica pediatra dermatóloga.
3. Médica dermatóloga.
4. Médico parasitólogo. Posgrado de dermatología.
5. Profesora de dermatología.
Policlínica de Dermatología Pediátrica. Centro Hospitalario Pereira Rossell.
Fecha recibido: 24 de mayo de 2004
Fecha aprobado: 28 de diciembre de 2004
Resumen
Se presenta el caso clínico de una paciente de 18 meses que consulta en policlínica de dermatología pediátrica por lesiones interpretadas como calcinosis cutánea.
Se analizan las causas de calcinosis cutánea, llegando al diagnóstico de osteodistrofia hereditaria de Albright.
Se describen las características clínicas y diagnósticas de esta enfermedad.
Palabras clave: CALCINOSIS-etiología
CALCINOSIS-diagnóstico
PIEL-patología
Summary
The objective of this paper is to show the clinic case of eighteen months patient who consults, at pediatric dermatology polyclinic, for lesions interpreted how calcification of the skin.
The causes of calcification are analyzed; the diagnostic is Albright’s hereditary osteodystrophy.
The clinics and diagnostics characteristics of that pathology are described.
Key words: CALCINOSIS-etiology
CALCINOSIS-diagnosis
SKIN-pathology
Introducción
La calcinosis cutánea o calcinosis cutis es el depósito de sales de calcio insolubles en tejidos cutáneos (1).
Si bien la presencia de una lesión calcificada en la piel no constituye un motivo de consulta frecuente en dermatología pediátrica, es necesario contar con un enfoque que nos permita orientarnos frente a ésta desde el punto de vista etiológico, así como decidir los estudios paraclínicos a solicitar.
Objetivos
- Describir las causas de calcinosis cutánea.
- Mostrar cómo a partir de una lesión en piel en apariencia banal, se puede llegar al diagnóstico de una patología endocrinológica.
- Describir las características clínicas y paraclínicas de la osteodistrofia hereditaria de Albright.
Caso clínico
MD. Sexo femenino, raza blanca, procedente de Montevideo. Edad: 18 meses.
Motivo de consulta: enviada para valoración de lesiones en piel, localizadas en muslo derecho y región lumbar izquierda, de 3 y 2 centímetros de diámetro mayor respectivamente, eritemato–violáceas que presentan a la palpación nódulos de consistencia pétrea que se visualizan al comprimir la piel entre dos dedos (figura 1). Estas lesiones aparecieron a los seis meses de edad. No presentó episodios de tetania ni convulsiones.
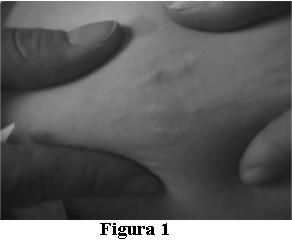
Se realizó diagnóstico clínico presuntivo de calcinosis cutánea.
Del resto del examen físico se destaca: paciente con cara de luna llena, raíz nasal deprimida, impresiona con acortamiento de los metacarpianos (figura 2), signos de Trousseau y Chvostek negativos, resto del examen normal.
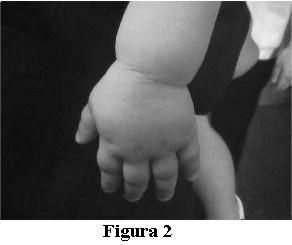
Antecedentes perinatales: primera gesta, embarazo bien controlado, mal tolerado por amenaza de aborto e infección urinaria. Rotura prematura de membranas a las 36 semanas. Parto vaginal. Recién nacido vigoroso, 2.600 g, 48,5 cm. Hipoglicemia a las 24 horas de vida. Alta a las 72 horas.
Crecimiento y desarrollo: obesidad desde los seis meses en tratamiento. Peso actual 14.300 kg (>p97), talla 80 cm, (p40). Camina con ayuda. Áreas sicomotriz, social y lenguaje normales.
Alimentación: pecho directo hasta los 11 meses. Hierro y vitamina D hasta hace un mes. Cumple plan de alimentación indicado en la policlínica de obesidad.
Antecedentes patológicos: bronquiolitis a los 6 meses. Luego episodios de broncoespasmo leves.
Hipotiroidismo subclínico diagnosticado a los 12 meses. TSH: 12, 62 mUI/ml (0,85-6,46), T3: 4,43 pg/ml (2,47-5,34), T4: 0,94 pg/ml (0,94-1,71). Recibe levotiroxina sódica.
CEV vigente.
Antecedentes familiares: madre portadora de patología siquiátrica. Varias internaciones por intentos de autoeliminación y depresión. Se desconocen antecedentes paternos.
La niña está a cargo de la abuela.
Medio socioeconómico y cultural aceptable.
Exámenes de laboratorio:
- Dosificación de PTH: 192 y 217 pg/ml (normal: 8–76 pg/ml).
- Calcemia: 9,7 mg/dl (normal: 8,8–10,8 mg/dl).
- Fósforo: 5,7 mg/dl (normal: 2,9–5,4 mg/dl).
- Radiografía de manos: acortamiento de metacarpianos.
- Radiografía de cráneo: normal
Se valoró la realización de biopsia cutánea que no se consideró necesaria.
Discusión
Las diferentes causas de calcinosis cutánea pueden ser divididas en cuatro categorías principales (1,2): distrófica, metastásica, iatrogénica e idiopática; así como también formando parte de entidades sindromáticas (tabla 1).

La calcificación distrófica es el tipo más común de calcinosis cutánea y ocurre como resultado de una lesión o anormalidad local de los tejidos, generalmente con metabolismo fosfocálcico normal. Esta categoría agrupa diversas enfermedades (enfermedades del tejido conectivo autoinmunes y congénitas, neoplasias, infecciones, etcétera).
El análisis de la historia clínica de esta paciente no nos orienta a ninguna de estas enfermedades. Dentro de las neoplasias cutáneas, si bien no se plantearon en este caso, merecería recordarse el pilomatricoma o epitelioma calcificante de Malherbe, por ser la que más comúnmente presenta calcificación y osificación, así como por ser frecuente su presentación en la niñez. Este es un tumor que se localiza en la dermis, con diferenciación hacia estructuras pilosas, que se presenta como un nódulo profundo de consistencia firme de 0,5-5 cm, cubierto por piel normal, localizado más frecuentemente en cara y extremidades superiores (3).
La calcificación metastásica consiste en la precipitación de sales de calcio en tejido normal como consecuencia de una alteración del metabolismo fosfocálcico. La calcificación puede estar muy extendida afectando vasos sanguíneos, riñones, pulmones y mucosa gástrica, así como la piel. En este grupo se incluyen el hiperparatiroidismo secundario de la insuficiencia renal crónica y la intoxicación por vitamina D, que deberá ser tenida en cuenta en la edad pediátrica, no planteables en este caso clínico.
La calcinosis cutánea de causa iatrogénica se produce como complicación de la terapia intravenosa con cloruro o gluconato de calcio.
Dentro de la categoría de la calcinosis idiopática, los nódulos subepidérmicos calcificados aparecen como una lesión solitaria de 3 a 11 mm en áreas expuestas de cabeza y extremidades, pueden ser congénitos o adquiridos. Algunos investigadores creen que estas lesiones representan hamartomas calcificados de glándulas sudoríparas (1,4). Existen cuatro síndromes osificantes bien descritos (1): la fibrodisplasia osificante progresiva; la heteroplasia ósea progresiva; el osteoma cutáneo en forma de placa y la osteodistrofia hereditaria de Albright.
La fibrodisplasia osificante progresiva presenta osificación progresiva del tejido conectivo profundo, así como dedos gordos de pies dismórficos (halux), elemento éste de utilidad para realizar un diagnóstico temprano y prevenir la aparición de nuevas lesiones calcificadas, ya que éstas se producen luego de traumatismos (4,5).
La heteroplasia ósea progresiva y el osteoma cutáneo en forma de placa son enfermedades que se pueden presentar durante la lactancia. En ambas la calcificación se inicia en la dermis y se expande progresivamente, en el primer caso hasta comprometer tejidos profundos como el músculo (1).
La osteodistrofia hereditaria de Albright (OHA) es una enfermedad genética, transmitida con carácter autosómico dominante con resistencia a la acción de la paratohormona (PTH) (pseudohipoparatiroidismo) (6-9). Los niños afectados presentan obesidad, cara redonda, estatura baja, acortamiento del 4º metacarpiano y otros huesos de los pies, manos y calcificaciones subcutáneas. En la evolución pueden agregar calcificaciones de ganglios basales y cataratas lenticulares. En esta niña, dada la presencia de obesidad con cara de luna llena, raíz nasal deprimida, braquidactilia y calcificaciones subcutáneas nos orientamos clínicamente a esta enfermedad. No se realizó estudio ocular para descartar existencia de cataratas.
Estos pacientes pueden asociar hipotiroidismo, frecuentemente subclínico, como en este caso.
El defecto subyacente es una anomalía en el receptor de PTH o en la transmisión de su señal que es mediada por el AMP cíclico. En ésta intervienen tres elementos: el receptor de PTH, el sistema de proteína G de la membrana y la adenilato ciclasa.
En el tipo 1a, que es el más frecuente, existe un defecto de la subunidad alfa de la proteína de la unión del nucleótido guanina (G5 alfa), que constituye un factor de acoplamiento necesario para que la PTH se una al receptor y active el AMP cíclico. El gen de la G5 alfa se localiza en el cromosoma 20q 13.2. El 50% de estos pacientes tienen retardo mental.
En el tipo 1b, la proteína G es normal pero el AMP cíclico no aumenta con el estímulo de la PTH. El tipo II muestra aumento del AMP cíclico, la respuesta del AMP cíclico tras la inyección intravenosa de PTH es normal. No se asocia a OHA. Además de resistencia a la PTH se puede observar resistencia a la tirotropina, gonadotropina y glucagón. El hipotiroidismo clínico no es frecuente, por el contrario sí lo es la disfunción gonadal. Los pacientes se pueden encontrar hipocalcémicos, pudiendo ser la tetania un signo de presentación, así como normocalcémicos al momento del diagnóstico desarrollando la alteración en la evolución.
Los niveles de fósforo están aumentados y los de vitamina D3 descendidos. Algunos pacientes pueden presentar las características fenotípicas de la OHA con niveles de calcio y fósforo normales y PTH normal o levemente aumentada. Esta entidad se conoce como pseudopseudohipoparatiroidismo.
Existen otras formas de pseudohipoparatiroidismo, como el tipo 1c que también pueden tener OHA. El tipo II no tiene OHA.
La variabilidad en la expresión clínica sugiere que el gen mutado es un gen improntado con expresividad variable según se herede del padre o la madre (7).
La paciente fue derivada a policlínica de endocrinología, planteándose el diagnóstico de pseudohipoparatiroidismo tipo 1a por frecuencia, la confirmación definitiva se logra dosificando la proteína G estimuladora (actividad alfa-Gs) que se encuentra disminuida, lo que no se realiza en nuestro medio.
En relación a las manifestaciones cutáneas, continuaron apareciendo lesiones calcificadas, iniciándose tratamiento con hidróxido de aluminio, con el que se busca disminuir la absorción de fosfato, evitando que el producto calcio x fósforo en suero alcance el umbral a partir del cual se depositan las sales de calcio en los tejidos. No aparecieron nuevas calcificaciones en la evolución.
Conclusiones
En este caso, la presencia de una lesión en piel sirvió como guía diagnóstica de una enfermedad endocrinológica en la cual el diagnóstico temprano es fundamental para evitar complicaciones metabólicas así como en otros casos para detectar el hipotiroidismo subclínico que requiere tratamiento de sustitución hormonal inmediato.
Esto nos recuerda la importancia del examen dermatológico y su correcta interpretación, ya que la piel y sus anexos pueden actuar como marcador de muy diversas enfermedades.
Bibliografía
1. Walsh JS, Fairley JA. Mineralización y osificación cutáneas. En: Fitzpatrick T.B, Freedberg I.M, Eisen A.Z, Wolff K, Austen K.F, Goldsmith L.A, et al. Dermatología en Medicina General. 5ªed. Buenos Aires: Médica Panamericana, 2001: 1934-40.
2. Nico MM, Bergonse FN. Subepidermal Calcified Nodule: Report of two cases and Review of the literature. Pediatr Dermatol 2001; 18(3): 227-9.
3. Hashimoto K, Lever WF. Tumores de los anexos cutáneos. En: Fitzpatrick TB, Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, et al. Dermatología en Medicina General. 5ªed. Buenos Aires: Médica Panamericana, 2001: 949-50.
4. Juzych LA, Nordby CA. Subepidermal Calcified Nodule. Pediatr Dermatol 2001; 18(3): 238-40.
5. Sferco A. Fibrodisplasia Osificante Progresiva: Pautas para su reconocimiento. Arch Argent Pediatr 2001; 99(3): 249-52.
6. Sobradillo B, Fernández Ramos C, Rica I. Hipoparatiroidismo. En: Pombo M, Audi L, Bergada C, Bueno M, Calzada R, Dieguez C, et al. Tratado de Endocrinología Pediátrica. 3ª ed. Madrid: Mc Graw Hill-Interamericana, 2002: 631-45.
7. Doyle AD, Di George AM. Pseudohipoparatiroidismo: Osteodistrofia Hereditaria de Albright. En: Behrman RE, Kliegman RM, Jenson HB. Nelson. Tratado de Pediatría. 16 ed. Madrid: Mc Graw- Hill, Interamericana, 2000: 1874-5.
8. Soriano Guillén L, Muñoz Calvo MT, Sánchez Prieto E, Martínez Martín, Pozo Román J, Argente Oliver J. Pseudohipoparatiroidismo en la infancia. Aspectos clínicos y terapéuticos. Rev Esp Pediatr 2001; 57(4): 365-9.
9. Bastida Eizaguirre M, Iturbe Ortiz de Urbina R, Arto Urzainqui MJ, Ezquerra Larreina R, Escalada San Martín J. Osteodistrofia hereditaria de Albright. Identificación de una mutación original en una familia. An Esp Pediatr 2001; 54: 598-600.
Correspondencia: Dra. Andrea Spremolla.
E-mail: aspremolla@hotmail.com

