










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.74 no.3 Montevideo ago. 2003
Bronquiectasias en la infancia
DRES. PATRICIA MURTAGH 1, HEBE GONZáLEZ PENA 1, CLAUDIO CASTAñOS 2,
MARíA CATALINA PINCHAK 3, MARIO GRENOVILLE 4, MATíAS OLEASTRO 5, MóNICA SIMINOVICH 6
Resumen
Las bronquiectasias son dilataciones permanentes de las vías aéreas segmentarias y subsegmentarias. En la mayoría de los pacientes, las infecciones necrotizantes del pulmón, únicas o recurrentes, son las principales responsables del daño inicial.
Los objetivos de este trabajo fueron identificar las entidades causales y caracterizar los hallazgos clínicos, radiológicos y tomográficos que posibilitan el diagnóstico.
En el período desde agosto de 1987 a diciembre de 2001 se examinaron las historias clínicas de 687 pacientes con diagnóstico de bronquiectasia atendidos en el Hospital de Pediatría Dr. J.P. Garrahan de Buenos Aires, Argentina.
Las causas de bronquiectasia encontradas con mayor frecuencia fueron: posinfección respiratoria aguda baja inespecífica (n=403), fibrosis quística (n=110) e inmunodeficiencias primarias (n=73).
Conclusiones: los signos y síntomas clínicos y los hallazgos radiológicos y tomográficos permitieron el diagnóstico de bronquiectasias. En esta serie, la infección respiratoria aguda baja inespecífica fue la causa más frecuente de bronquiectasias, siendo muy precoz su edad de comienzo. En este grupo hubo un alto porcentaje de niños con lesiones extensas y severas que desarrollaron enfermedad pulmonar grave con insuficiencia respiratoria crónica en etapas tempranas de la vida.
Palabras clave: BRONQUIECTASIA-etiología
BRONQUIECTASIA-diagnóstico
Summary
Bronchiectasia are permanent dilations of the segmentary and sub-segmentary airway. In most patients either single or recurrent necrotizing infections of the lung are the main cause of the initial damage to the tissue.
The objective of this publication was to identify the causal entities, characterize the clinical, radiological and tommographic findings that enable the diagnosis.
During the time period August 1987 and December 2001 the case histories of 687 patients with a diagnosis of bronchiectasia attending the "Dr. J. P. Garrahan" Paediatric Hospital in Buenos Aires, Argentina, were reviewed.
Acute, non-specific, lower respiratory infection was the most frequent cause (n=403) of bronchiectasia. Cystic fibrosis (n=110), and primary immunodeficiency (n=73) followed.
Conclusions: the clinical signs and symptoms as well as the radiological and tommographic findings allowed bronchientasia to be diagnosed. In this series acute non-specific lower respiratory infection was found to be the most frequent cause of bronchiectasia. The age at presentation was very early. In this group of patients an important number of children presenting with extense and severe tissue damage whent on to develop severe lung disease with chronic respiratory insufficiency at early stages of life.
Key words BRONCHIECTASIS-etiology
BRONCHIECTASIS-diagnosis
Introducción
Las bronquiectasias (BQT) son dilataciones permanentes de las vías aéreas segmentarias y subsegmentarias. Son poco frecuentes, con evolución a la cronicidad por la irreversibilidad de las lesiones.
En general, se acepta que las bronquiectasias son el resultado de una variedad de enfermedades o de injurias pulmonares. En la mayoría de los pacientes, las infecciones necrotizantes del pulmón, únicas o recurrentes, son la causa más común del daño inicial. En muchos pacientes pediátricos, los síntomas ocurren luego de una infección respiratoria aguda baja en etapas tempranas de la vida.
Barker y Bardama (1) la denominaron enfermedad huérfana", implicando que es una enfermedad infrecuente que ha sido olvidada en el desarrollo de investigaciones en cuanto a su etiopatogenia y tratamiento. Sin embargo existe interés médico en el tema, ya que una búsqueda bibliográfica realizada entre 1966 y 2001 reveló la existencia de 1.807 publicaciones sobre bronquiectasias en la infancia.
En nuestro medio, como en otros países en vías de desarrollo, la infección respiratoria aguda baja es una de las principales causas de morbimortalidad en niños pequeños y un importante factor de riesgo para el desarrollo de enfermedad pulmonar crónica no sólo en la infancia (2-4) sino también en etapas posteriores de la vida (5).
La prevalencia de BQT en el mundo es desconocida. Hay publicaciones que indican una alta prevalencia en poblaciones aisladas con pobre acceso a centros asistenciales y alta frecuencia de infecciones del tracto respiratorio durante la infancia, como los nativos de Alaska en Yukón, Kuskokwim Delta (3).
Material y método
Población
Se analizaron retrospectivamente las historias clínicas de 687 pacientes atendidos en el Hospital de Pediatría Dr. Juan P. Garrahan de Buenos Aires durante un período de 14 años (agosto de 1987 a diciembre de 2001) con diagnóstico de BQT. Se excluyeron de esta revisión los pacientes con síndrome de inmunodeficiencia adquirida (SIDA) y tuberculosis activa.
El diagnóstico de BQT se realizó basándose en el hallazgo de tres o más signos o síntomas clínicos asociados a dos o más signos radiológicos.
En la evaluación clínica se destacó la importancia de la presencia de tos crónica o intermitente, broncorrea persistente, hemoptisis y dolor torácico. Al examen físico se consignó la auscultación persistente de subcrepitantes, respiración soplante y/o broncofonía y signos de enfermedad pulmonar crónica (retraso de crecimiento, aumento del diámetro ántero posterior del tórax, rigidez torácica, hipocratismo digital).
El retraso de crecimiento se evaluó a través del estudio antropométrico, que se realizó en forma estandarizada de acuerdo a normas previamente establecidas (6).
Se registró edad de inicio de los síntomas, antecedentes de infecciones respiratorias, asma, síndrome aspirativo crónico, presencia de enfermedades genéticas o congénitas y de otras patologías de base.
Para la identificación de las intercurrencias se siguieron los criterios definidos por O’Donnell y colaboradores (7), quienes consideraron la presencia de cuatro o más de los siguientes signos y síntomas: fiebre, aumento de tos y producción de esputo, dificultad respiratoria o disnea (de acuerdo a la edad), sibilancias, menor tolerancia al ejercicio y reducción de la función pulmonar. Asimismo la aparición de imágenes pulmonares nuevas en las radiografías de tórax y cambios en los ruidos respiratorios a la auscultación.
Estudio de imágenes
Todos los estudios fueron realizados en el Servicio de Radiología del hospital Garrahan.
Las radiografías de tórax de frente y perfil se realizaron con aparatos de rayos General Electric MPX y MST 1050 II. Se consideraron los criterios radiológicos de Woodring (8) para el diagnóstico de BQT: engrosamiento o pérdida de definición del intersticio peribroncovascular, dilatación bronquial con manguito peribrónquico, aproximación del intersticio peribroncovascular, pérdida de volumen de un segmento o lóbulo, imagen en anillo de sello, hiperinsuflación compensadora e imágenes quísticas. Se realizaron tomografías computadas de alta resolución (HRCT) (tomógrafo computado Toshiba 600 S de tercera generación) con cortes finos de 4 mm de espesor a intervalos de 5 mm. Las tomografías se realizaron sin anestesia y fuera de los episodios agudos. Los hallazgos tomográficos típicos de BQT fueron: evidencia de dilatación bronquial con el diámetro interno del bronquio mayor al diámetro de la arteria bronquial acompañante, dilataciones cilíndricas, varicoso-saculares y quísticas, engrosamiento del intersticio peribronquial, atelectasia lobar o segmentaria, atrapamiento aéreo, oligohemia en mosaico, hiperinsuflación compensadora y panalización.
Broncoscopía
El examen endoscópico de las vías aéreas y los aspirados bronquiales fueron realizados en el servicio de endoscopía respiratoria del hospital Garrahan (broncoscopios rígidos Pilling y broncofibroscopio Pentax). Se siguieron las indicaciones sugeridas por Barker y Bardana (1), quienes identificaron tres situaciones en las cuales la broncoscopía es de particular ayuda: a) cuando el diagnóstico no está aclarado y no se ha descartado definitivamente un cuerpo extraño o tumor endobronquial; b) cuando el diagnóstico de BQT ha sido establecido clínica y radiológicamente pero se sospecha que un segmento o lóbulo contribuye principalmente en la mayor producción de secreciones; c) cuando es necesario identificar el segmento o lóbulo responsable en caso de hemoptisis.
En ningún paciente se realizó broncografía.
Microbiología
En los pacientes atendidos en la etapa aguda en nuestro hospital se realizó el diagnóstico rápido para virus respiratorios por medio de la inmunofluorescencia indirecta (IFI) en aspirado de secreciones nasofaríngeas (Light Diagnostics - Chemicon International).
Las muestras de esputo o aspirado bronquial, o ambas, obtenidas en pacientes que no estuvieron recibiendo antibióticos 48 horas antes de este examen, fueron procesadas para gérmenes comunes con antibiograma en el laboratorio de microbiología de nuestro hospital según técnicas estándar.
Examen funcional respiratorio (EFR)
Las pruebas de función pulmonar (PFP) fueron realizadas en el laboratorio pulmonar del servicio de neumonología del hospital Garrahan. Para su realización los pacientes no debían haber padecido intercurrencias respiratorias agudas en las seis semanas previas al estudio.
En los niños mayores de seis años se realizaron espirometrías y curvas de flujo-volumen computarizadas pre y post inhalación de broncodilatadores ß2-adrenérgicos, consignándose la capacidad vital forzada (CVF), flujo espiratorio forzado en un segundo (VEF1), relación VEF1/CVF, flujo espiratorio medio forzado (FEF 25-75) (espirómetro Vitalograph 2170 Spirotrac IV). Se utilizaron las tablas de valores normales de Polgar (9), siguiendo las recomendaciones de la American Thoracic Society para estandarización de espirometrías (10), realizando los pacientes esta prueba en posición de pie. Se consideró patrón funcional de restricción a la disminución de la CVF menor del 80%. Se consideró patrón funcional de obstrucción a la presencia de un VEF1 menor del 80% asociado a una relación VEF1/CVF menor del 83%.
En los lactantes y niños pequeños entre 3-36 meses se efectuó examen funcional por análisis de la curva parcial de flujo volumen forzada por el método de compresión tóraco-abdominal rápida. El parámetro analizado fue el flujo máximo en la capacidad funcional residual (Vmax/FRC) (11). Estos estudios funcionales fueron realizados en el Laboratorio de Estudios Funcionales Respiratorios en Lactantes del Hospital Francés de Buenos Aires.
Diagnóstico de FQ y estudio genético
A todos los pacientes con BQT se les realizó al menos dos tests del sudor (Método de Gibson y Cooke).
La confirmación del diagnóstico de fibrosis quística (FQ) se basó en la existencia de dos tests del sudor positivos (sodio mayor a 60 mEq/L) o el hallazgo de dos mutaciones conocidas del gen de la FQ oambos.
En todos los pacientes con FQ se realizó estudio molecular. Luego de obtener el consentimiento informado, el análisis de las mutaciones del gen fue realizado en el laboratorio de biología molecular de nuestro hospital. Se estudiaron 13 mutaciones.
Con el estudio conjunto de la DF 508, G542X, W1282X, N1303K, 3849+10KvC®T, R334W®T, se logró caracterizar el 70,5% de los alelos fibroquísticos de los pacientes (12).
Estudio inmunológico
Para estudiar el estado de inmunodeficiencia se realizó: hemograma completo con recuento y fórmula leucocitaria, dosaje de inmunoglobulinas séricas IgG, IgA e IgM por nefelometría (Beckman Array 360), dosaje de subclases de Ig G sérica por ELISA (antisueros de ratón contra IgG1, IgG2, IgG3, IgG4, Sigma), título de isohemaglutininas, producción de anticuerpos específicos postvaccinales frente a antígeno proteico toxoide tetánico por hemaglutinación y/o ELISA (con antígeno purificado, Statem Serum Institud, Denmark) y frente a antígenos polisacáridos de Streptococcus pneumoniae por ELISA (Binding Site, Birmingham, UK), niveles de IgE sérica por ELISA (ABBOTT), inmunomarcación linfocitaria en muestras de sangre periférica para linfocitos por citometría de flujo (FACSort, Beckton Dickinson) con anticuerpos monoclonales (Beckton Dickinson) contra T (CD 3, CD 4, CD 8), linfocitos B (CD 19) y linfocitos NK (CD 16-56), técnicas de biología molecular como SSCP (Single-strand conformational polymorphism) y secuenciación directa fueron realizadas para determinar la presencia de mutaciones en el gen btk con el fin de diferenciar aquellos casos de varones con agama/hipogamaglobulinemia y ausencia de linfocitos B. Todos los resultados obtenidos se compararon con valores de referencia para la edad.
Se consideró déficit de algunas de las subclases cuando las concentraciones se ubicaron repetidas veces por debajo del percentil 5 de los valores referidos para la edad.
Las inmunodeficiencias fueron agrupadas de acuerdo a la recomendación propuesta por el grupo de expertos de la OMS (13).
Cirugía
Para la indicación quirúrgica se siguieron los criterios definidos por Wilson y Deber (14) que son: enfermedad localizada con sintomatología importante que interfiera con una vida normal, riesgo de hemoptisis grave en un área identificable, enfermedad localizada con retraso del crecimiento, enfermedad localizada con infecciones recurrentes, enfermedad inestable asociada a significativa progresión y/o extensión de BQT resecables, BQT no fácilmente ni totalmente resecables pero con síntomas que impliquen riesgo de vida o invalidez.
Examen anatomopatológico
Las piezas quirúrgicas obtenidas fueron fijadas en formol al 10%. Se tomaron cortes representativos que fueron incluidos en parafina y coloreados con Hematoxilina-Eosina, PAS, Tricrómico de Masson y Van Giesson para fibras elásticas.
Resultados
La incidencia de BQT en nuestro hospital fue de 2,27/10.000 consultas anuales, incluyendo a los niños con fibrosis quística y de 1,93/10.000 no considerando a esta enfermedad.
Pertenecieron al sexo masculino 385 pacientes (56%).
De acuerdo a su etiología, las BQT se clasificaron en cuatro grandes grupos: posinfecciosas, secundarias a aspiración, secundarias a enfermedades hereditarias o congénitas y un grupo de misceláneas (tabla 1).
En 28 pacientes el diagnóstico fue anatomopatológico: 21 fueron lóbulos resecados por malformaciones pulmonares (15 fueron malformaciones adenomatoideas quísticas (MAQ), cuatro secuestros pulmonares intralobares y dos hipoplasias pulmonares). En dos pacientes con tumores pseudoinflamatorios pulmonares y en un paciente lobectomizado por una fístula broncopleural crónica y supuración pleuropulmonar se diagnosticaron BQT. Cuatro pacientes fueron operados con el diagnóstico de MAQ y resultaron ser BQT quísticas.
Analizaremos en particular los tres grupos más frecuentemente diagnosticados:
1) BQT secundarias a infecciones respiratorias bajas específicas.
2) BQT secundarias a fibrosis quística (FQ).
3) BQT secundarias a inmunodeficiencias primarias (IDP).
BQT secundarias a infección respiratoria aguda baja
La causa más frecuente en nuestro estudio correspondió a este grupo etiológico (n: 403; 58,6%).
El 89% de los niños eran sanos en el momento de la injuria inicial y 76% tenían menos de dos años. La neumonía multifocal (53,3%) fue la causa más frecuente; los agentes etiológicos causales de la IRAB fueron adenovirus en 25,3% y en siete pacientes se realizó la serotipificación, encontrándose el serotipo Ad7h reconocido por su gran agresividad.
La radiografía demostró un amplio predominio de lesiones bilaterales (73%), la localización más frecuente de las BQT fue en los lóbulos inferiores, en 83% de los casos se hallaba comprometido más de un lóbulo.
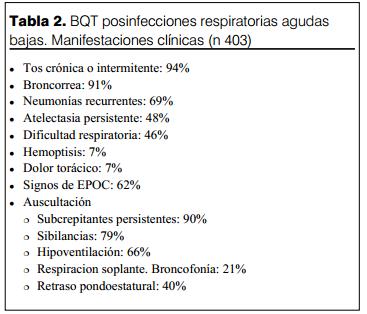
Los síntomas y signos encontrados se describen en la tabla 2.
Del análisis de 394 radiografías se confirmaron los criterios de Woodring y colaboradores (8) y se agregaron otros: reticulado alveolar, aproximación del intersticio peribroncovascular ("imágenes amontonadas") (Fraser y Paré, 1979) (15) y panalización (figuras 1 y 2).
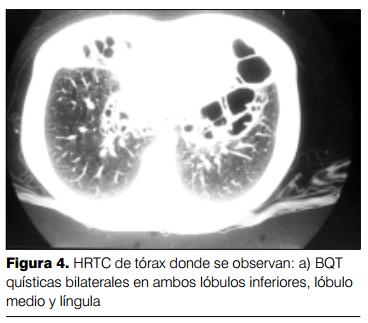
El análisis de 238 tomografías computadas (HRCT) permitió no solo confirmar el diagnóstico de BQT sino evaluar la extensión y el tipo de compromiso del parénquima pulmonar y alteraciones de la vascularización con el hallazgo de un alto porcentaje de oligohemia en mosaico (34%).
Las anormalidades específicas encontradas fueron: dilataciones cilíndricas, varicososaculares y quísticas (100%), engrosamiento del intersticio peribronquial (70%), atelectasia lobar o segmentaria (66%), atrapamiento aéreo (39%), oligohemia en mosaico (34%), hiperinsuflación compensadora (21%) y panalización (2%) (figuras 3 y 4).
En niños mayores de 6 años se realizaron 184 espirometrías y curvas de flujo–volumen computarizados. Los resultados demostraron exámenes funcionales normales en 36% de los casos, incapacidad ventilatoria obstructiva con respuesta variable luego de uso de broncodilatadores B2 agonistas en el 27%, incapacidad ventilatoria mixta en 21% y con incapacidad ventilatoria restrictiva en 16%. En estos dos últimos trastornos funcionales el grado de compromiso estuvo en relación directa con el número de lóbulos afectados. En 49 niños pequeños los estudios funcionales pusieron en evidencia una importante caída en los flujos espiratorios máximos a capacidad residual funcional (Vmax/FRC) con escasa o nula respuesta luego del uso de broncodilatadores.
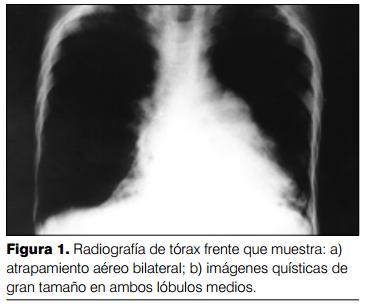
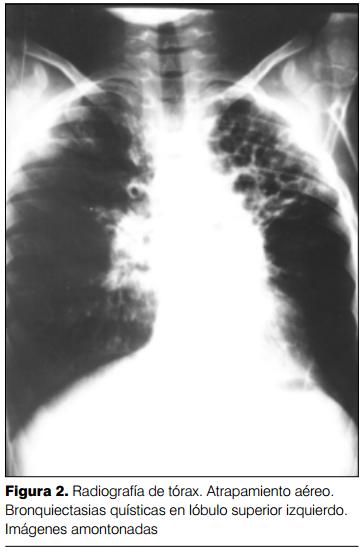
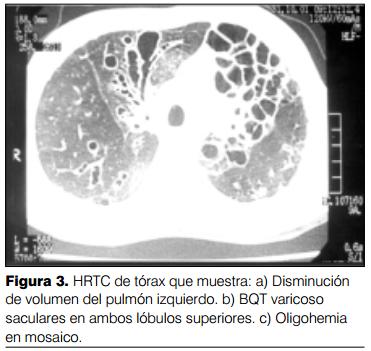
Los signos endoscópicos encontrados fueron: aumento de secreciones purulentas y espolones bronquiales ensanchados con mucosa bronquial congestiva o sangrante.
En el estudio bacteriológico de los aspirados bronquiales y esputos (n 296) se observó una alta contaminación de flora polimicrobiana de la zona orofaríngea. El germen más frecuente hallado como patógeno único o formando parte de varias asociaciones fue el Haemophylus influenzae b (18,4%), con 2% del total de cepas productoras de b-lactamasa, y en 6,9% el Streptococcus pneumoniae sensible a amoxicilina, quedando sin identificar gérmenes en 12,4% de los casos.
En nuestro estudio se realizaron 58 cirugías. Las indicaciones fueron atelectasia crónica (n=28), atelectasia crónica y hemoptisis (n=10), hemoptisis (n=5), malformación adenomatoidea quística (n=4), BQT quísticas localizadas con infección recurrente (n=6), infección recurrente localizada (n=4), fístula broncopleural crónica en una supuración pleuropulmonar (n=1).
Se realizó el estudio anatomopatológico en el material obtenido de los 58 pacientes.
Las lesiones se localizaron en el pulmón izquierdo en 38 casos (65,5%). Las piezas correspondieron a 41 lobectomías, cuatro bilobectomías, una lobectomía atípica, siete lobectomías y segmentectomías de lóbulos adyacentes y ocho neumonectomías.
BQT secundarias a fibrosis quística (FQ)
Desde el año 1988 se han diagnosticado en este hospital 220 pacientes con FQ, de los cuales 160 ingresaron a un programa de seguimiento. Veinte fallecieron (12,5%). Un total de 110 presentaron bronquiectasias al momento del estudio. Los gérmenes más frecuentemente encontrados en los cultivos de secreciones bronquiales fueron: Pseudomona aeruginosa como germen único en 46 pacientes (41,8%) y Pseudomona aeruginosa y Staphylococcus aureus en 30 pacientes (27,2%). De éstos, cuatro pacientes presentaron Staphylococcus aureus meticilino resistente.
El Staphylococcus aureus se presentó como germen único en 26 pacientes (23,6%). El Haemophylus influenzae b se presentó en un paciente, al igual que la Burkholderia cepacia y la Stenotrophomona maltophilia.
En todos los pacientes se realizó estudio genético. Se pudieron detectar 39 mutaciones del gen de la FQ, incluyendo cinco mutaciones que no habían sido descritas anteriormente y dos polimorfismos nuevos. La mutación más frecuentemente encontrada fue la DF508, encontrándose en 57% de los alelos analizados.
De los pacientes que tenían BQT, 37,5% fueron homocigotos con DF508 y 25% heterocigotos, lo cual indica para nuestra población la presencia de esta mutación en 62,5% de los pacientes. Las otras mutaciones que se encontraron fueron: G542X (7,9%), N1303K (4,5%), 3849+10-kbcT (0,02%), W1282x (4,5%) y otras menos frecuentes 1717-1G-A, 621+1GA y DI507.
BQT en inmunodeficiencias primarias
Del total de 687 pacientes con BQT, 73 casos (39 varones) presentaron una inmunodeficiencia primaria como enfermedad predisponente. La edad media de esta población al momento de la evaluación fue de 13 años (rango 4-25 años).
Las deficiencias predominantemente de anticuerpos fueron las más frecuentes, con 52 casos (71,2%). Dentro de ellas, la inmunodeficiencia común variable y la agamaglobulinemia ligada al sexo representaron el 46,5% del total de casos con 21 y 13 casos respectivamente. Siguieron en orden de frecuencia la deficiencia selectiva de IgA y la deficiencia de anticuerpos con inmunoglobulinas normales con ocho casos cada una. La deficiencia de la subclase de IgG2 fue confirmada en dos casos.
Las IDP agrupadas como otros síndromes de ID bien definidos fueron las que siguieron en frecuencia a aquellas deficiencias de anticuerpos. Este grupo fue representado por el síndrome de hiper-IgE con cinco casos, síndrome de Wiskott-Aldrich con dos casos y candidiasis cutáneo mucosa crónica en un caso.
En tercer lugar se ubicaron los síndromes asociados a ruptura de ADN con cinco casos de ataxia-telangiectasia y un caso con síndrome de Nijmegen. Los defectos del fagocito sólo fueron diagnosticados en dos oportunidades como síndrome de Kostmann. Las deficiencias combinadas sólo se vieron representadas en un caso de deficiencia de exposición de moléculas de HLA clase II.
Dos pacientes presentaron inmunodeficiencia asociada a un cuadro de hipoplasia cartílago pelo y, por último, dos casos no pudieron ser caracterizados.
La edad media al momento del diagnóstico de la IDP fue de 6,23 años (rango 0,57–16,16) mientras que la de diagnóstico neumológico de BQT fue de 7,30 (rango 0,41–16,16).
La edad media en la cual los pacientes iniciaron sus manifestaciones respiratorias fue de dos años (rango 0,12-8). El dosaje de inmunoglobulinas séricas IgG, IgA, IgM, e IgE orientó o confirmó el diagnóstico en 47 casos (64,3%). En los restantes 26 casos fue necesario contar con estudios más complejos para definir el verdadero compromiso inmunitario. Nueve pacientes de este grupo fallecieron y en tres de ellos la muerte fue consecuencia de la enfermedad pulmonar crónica.
Discusión
No existen estimaciones adecuadas de la real incidencia y prevalencia de las BQT en los países en desarrollo. Los estudios epidemiológicos de países industrializados son antiguos. Field y colaboradores en 1969 (17) analizaron los índices de admisiones anuales por BQT en cinco hospitales de Gran Bretaña entre 1938 y 1961, y notaron una disminución drástica en las admisiones pediátricas desde una media de 48/10.000 en 1952 a 10/10.000 en 1960. Atribuyeron este descenso al uso de antibióticos de amplio espectro, a las inmunizaciones activas para el control de enfermedades tales como el sarampión y coqueluche y al control de la tuberculosis.
Otra cita bibliográfica refiere una incidencia posneumonía de 1,06/10.000 (Clark, 1963) (18). La baja prevalencia en países desarrollados ha sido reducida por los factores ya mencionados, y en estos países las principales causas de BQT son debidas a fibrosis quística e inmunodeficiencias. No ocurre lo mismo en los países en desarrollo, donde a pesar de su subdiagnóstico, las BQT constituyen un grupo importante de la patología pulmonar crónica. De acuerdo con nuestros datos, la prevalencia registrada en nuestro hospital de BQT post IRAB es de 1,34/10.000 consultas; no parece ser alta comparada con datos de países desarrollados de hace 40 años atrás, pero hemos excluido otras causas posinfecciosas de BQT, como sarampión, coqueluche y tuberculosis que eran consideradas en las estadísticas mencionadas. La importancia de estas infecciones respiratorias específicas no tiene significación en el número total de nuestros casos. Además, en nuestro medio se registra un amplio uso de antibióticos para el tratamiento de las IRAB. O sea que los mismos factores que disminuyeron la incidencia en países desarrollados deberían haber actuado en nuestra área. Sin embargo, cuando uno considera los porcentajes de BQT post IRAB la diferencia se hace notable, ya que representan casi 60% de los casos diagnosticados en nuestro estudio.
No existen trabajos recientes con un número importante de BQT en pacientes pediátricos de países desarrollados, por lo que hemos considerado para la comparación de nuestros hallazgos tres trabajos con los datos de más de 100 pacientes pediátricos que fueron publicados hace más de 35 años: Clark (1963, n 116) (18), Strang (1956, n 209) (19) y Glauser (1966, n 187) (20).
En nuestros pacientes la lesión pulmonar fue mucho más extensa, con mayor número de lóbulos comprometidos y se presentó a edades más tempranas. Nuestra revisión demostró mayor número de niños con dificultad respiratoria, sibilancias recurrentes y enfermedad pulmonar crónica con hipoxemia. El 32% del total de niños post IRAB presentó insuficiencia respiratoria crónica requiriendo O2 suplementario por períodos prolongados. La frecuencia de las otras manifestaciones clínicas de nuestros pacientes, como tos crónica, broncorrea, hemoptisis fueron similares a las referidas por estos autores.
Las sibilancias recurrentes se diagnosticaron especialmente cuando existía compromiso multilobar. Un signo clínico de alta sensibilidad y fácil diagnóstico semiológico fue el hallazgo de estertores subcrepitantes persistentes localizados en las zonas afectadas
En cuanto a la patogenia de la enfermedad inicial estamos convencidos de que por la edad de presentación, la evolución clínica y radiológica durante la etapa aguda del proceso, la evolución de algunos pacientes hacia la bronquiolitis obliterante asociada a la escasa o nula respuesta a los antibióticos de amplio espectro utilizados, la mayor parte de estos niños padecieron una infección pulmonar inicial de etiología viral. Las dificultades para el diagnóstico etiológico de las BQT post IRAB están vinculadas a la imposibilidad de estudio virológico de rutina y a la subestimación de la importancia de la etiología viral en las formas graves de IRAB.
A pesar de este subdiagnóstico, en el 25,3 % de los casos la infección inicial pudo ser relacionada con infección por adenovirus, agente que es endémico en nuestro medio.
Con respecto a los hallazgos bacteriológicos es conveniente estar alerta sobre la aparición de cepas productoras de b-lactamasas cuando los niños están colonizados por Haemophylus influenzae b.
El tratamiento se realizó de acuerdo al resultado bacteriológico del esputo o aspirado bronquial y antibiograma. Las dosis fueron las habituales, se indicaron ciclos no menores a 15 días. En pacientes con infecciones no controladas, la administración de los antibióticos se prolongó durante varios meses. En los pacientes inmunocompetentes no se indicó profilaxis con dosis únicas de antibióticos. El inicio temprano de la antibioticoterapia en las exacerbaciones de pacientes con BQT podría limitar el círculo vicioso de infección-inflamación. Se utilizaron broncodilatadores b2 adrenérgicos asociados o no a bromuro de ipratropium, según el resultado de la respuesta espirométrica. Como antinflamatorios se utilizaron corticoides tópicos en aerosol a dosis habituales (beclometasona, budesonide, fluticasona). La administración de los broncodilatadores fue previa a la kinesioterapia y los corticoides tópicos se administraron luego de finalizada la misma, para lograr una mayor efectividad. A todos los pacientes se les indicó tratamiento kinésico y drenaje postural, además de las maniobras convencionales para la movilización de secreciones bronquiales. A los niños mayores se los adiestró con diferentes técnicas respiratorias: ciclo activo de la respiración, control respiratorio, ejercicios de expansión torácica, aspiración forzada (HUFF) y drenaje autogénico y dispositivos mecánicos como el flutter, generando de esa manera una presión positiva oscilatoria en la vía aérea. Todas estas maniobras mejoran la eliminación de secreciones.
En aquellos pacientes con hipoxemia crónica (saturaciones de O2 inferiores a 92%) se indicó oxigenoterapia domiciliaria y fueron controlados en un programa especial para el seguimiento de estos pacientes.
Destacamos el enorme valor que le damos a la HRCT que en pediatría ha reemplazado a la broncografía para la confirmación diagnóstica de BQT. Su valor ya ha sido destacado por varios autores (21,22). El análisis de las 238 HRCT nos permitió no sólo confirmar el diagnóstico de BQT, sino además evaluar la extensión y el tipo de compromiso del resto del parénquima pulmonar y alteraciones de la vascularización con el hallazgo de un alto porcentaje de "oligohemia en mosaico" (34%), que ha sido descrita como patognomónica de bronquiolitis obliterante.
La realización de este estudio debe indicarse fuera de los períodos de exacerbaciones de la enfermedad y en niños mayores de cuatro años. A esta edad, el estudio se puede efectuar sin anestesia evitando los artefactos producidos por la hipoventilación de las zonas pulmonares dependientes.
Su indicación es fundamental en los pacientes en los que se plantee tratamiento quirúrgico de las BQT.
En nuestra casuística la FQ fue la segunda causa más frecuente de BQT.
Es la enfermedad genética letal más frecuente en la raza blanca, autonómica recesiva, con una frecuencia en la población de 1/3.000 recién nacidos vivos.
La FQ es una enfermedad crónica multisistémica que se caracteriza por presentar un defecto en el transporte iónico en la membrana epitelial de diferentes órganos, siendo los más afectados el sistema respiratorio, el digestivo y el reproductor.
La morbilidad predominante está dada por la enfermedad pulmonar obstructiva progresiva. Las primeras descripciones de la enfermedad fueron realizadas por Andersen en 1938 (23). En la década de 1980 se estableció que el defecto fisiológico fundamental es la falla en un canal de cloro regulado por AMPc (24-26).
En 1985 se describió que la mutación responsable del defecto está localizada en el cromosoma 7 (27). En el año 1989 se logra la clonación del gen (28-30), a partir de este momento se conoce la estructura y función de la proteína que es producida por el gen de la FQ, llamada CFTR (cystic fibrosis transmembrane conductance regulator).
El estudio molecular del gen CFTR provee diagnóstico temprano y de certeza a un gran número de pacientes, y es especialmente importante en los casos de test del sudor negativo. Posibilita asesoramiento genético a las familias e información necesaria para acceder a nuevos tratamientos
Los gérmenes más frecuentes hallados en el esputo de estos pacientes son el Staphylococcus aureus, el Haemophylus influenzae y la Pseudomona aeruginosa ya sean en sus formas mucosas como no mucosas.
Recientemente se ha explicado porqué la alta colonización con Pseudomona aeruginosa en estos pacientes, ya que otras enfermedades respiratorias (síndrome de cilias inmóviles y síndrome de Kartagener) que se comportan de forma parecida no presentan colonización con estos gérmenes.
En el año 1996 se describió la presencia de sustancias con poder bactericida que estuvieran inhibidas por la alta concentración de cloruro de sodio en el epitelio bronquial (31).
En el año 1997 se reportó una sustancia denominada B-Defensina humana, tipo I, que es un producto proteico con poder bactericida para la Pseudomona aeruginosa que es inactivada por las altas concentraciones de cloruro de sodio (32).
La infección e inflamación produce una enfermedad pulmonar obstructiva progresiva con BQT e insuficiencia respiratoria.
Dado que la FQ es una enfermedad progresiva y letal, la posibilidad de desarrollar BQT depende del grado de compromiso pulmonar y de la edad del paciente.
Las IDP constituyen un grupo heterogéneo de enfermedades, en su gran mayoría determinadas genéticamente, cuya característica común es la de presentar gran susceptibilidad para desarrollar infecciones.
La deficiencia puede involucrar cualquier componente del sistema inmune. Son enfermedades poco frecuentes cuya verdadera incidencia se desconoce aunque puede estimarse en 1 cada 10.000 nacidos vivos (33).
Las infecciones pueden ser tórpidas, recurrentes, crónicas y/o severas por microorganismos comunes u oportunistas. Aunque pueden presentarse en cualquier localización, el compromiso del aparato respiratorio en forma de sinusitis, otitis, bronquitis o neumonías es lo habitual (34,35). En aquellos casos con deficiente producción de anticuerpos, la terapia sustitutiva con gamaglobulina endovenosa (GGEV) ha demostrado su beneficio reduciendo la frecuencia y la severidad de las infecciones a nivel pulmonar (36,37), demostrado en nuestro estudio con la evolución de ocho pacientes (36,3%). Sin embargo, dos pacientes igual desarrollaron BQT aun recibiendo GGEV en forma adecuada, demostrando que la deficiente producción local de anticuerpos no sería siempre eficazmente compensada por el aporte parenteral de gamaglobulina. Si bien esta terapéutica no modifica la inflamación local, sí lo hacen otras medidas como la kinesioterapia y el drenaje postural.
En 17 casos (23,2%) el diagnóstico de IDP fue posterior al de BQT, esto demuestra un bajo índice de sospecha de IDP de base en pacientes con infecciones respiratorias recurrentes o crónicas.
El análisis del compromiso inmune demostró que el diagnóstico se confirmó en 64% con estudios de primera línea diagnóstica, demostrando la necesidad de estudios más complejos en un alto porcentaje de pacientes.
Esta enfermedad tiene alta morbilidad y en algunos casos su severidad tiene un alto impacto en la calidad de vida de los niños. Es necesario estar alerta frente a pacientes con BQT e infecciones recurrentes y descartar la IDF como etiología de base para instaurar una terapéutica precoz y consejo genético.

Conclusiones
La infección respiratoria aguda baja fue la causa más frecuente de BQT, destacando su inicio en edades muy tempranas de la vida, con lesiones bilaterales y desarrollo de enfermedad pulmonar e insuficiencia respiratoria crónica en alto número de pacientes.
Destacamos la importancia de la tomografía computada de alta resolución para el diagnóstico y para evaluar las alteraciones del parénquima pulmonar circundante.
El reconocimiento precoz de las exacerbaciones y su tratamiento adecuado acortan el círculo vicioso y mejoran la calidad de vida de estos pacientes.
La identificación de inmunodeficiencias y de fibrosis quística tiene importantes implicancias terapeúticas y pronósticas por lo cual frente al diagnóstico de BQT es necesario realizar estudios exhaustivos para descartar dichas enfermedades.
Agradecimientos
Agradecemos a los siguientes servicios sin cuya valiosa colaboracion este trabajo no hubiera podido ser realizado:
- Servicio de Imágenes. Hospital de Pediatría Garrahan.
- Servicio de Endoscopía. Hospital de Pediatría Garrahan.
- Servicio de Cirugía. Hospital de Pediatría Garrahan.
- Servicio de Microbiología. Hospital de Pediatría Garrahan.
- Servicio de Kinesiología. Hospital de Pediatría Garrahan.
- Servicio de Inmunología. Hospital de Pediatría Garrahan.
- Laboratorio de Biología Molecular. Hospital de Pediatría Garrahan.
- Laboratorio de Neumonología Pediátrica. Hospital Francés de Buenos Aires.
- Departamento de Estadística. Hospital de Pediatría Garrahan.
- Archivo Central de Historias Clínicas. Hospital de Pediatría Garrahan.
- Al Dr. Eduardo Diez (médico radiólogo pediatra) por su asesoramiento en la revisión de las imágenes radiológicas y tomográficas.
- Y a todos los médicos, enfermeras y personal de secretaría que atendieron con tanta dedicación y cariño a todos estos pacientes.
Bibliografía
1. Barker A, Bardana E (Jr). Bronchiectasis: update of an orphan disease (State of the Art). Am Rev Respir Dis 1998; 137: 969-78.
2. Bauer G, Leske V, Del Pino M, Keklikian E. Enfermedad Pulmonar obstructiva crónica postviral (EPOC). Resumen 650. Procedente del 31º Congreso Argentino de Pediatria 1997, Set. 24-27, Mendoza, Argentina.
3. Singleton R, Morris A, Redding G, Poll J, Holck P, Martinez P et al. Bronchiectasis in Alaska native children: causes and clinical courses. Pediatr Pulmonol 2000; 29: 182-7.
4. Karakoc G, Yilmaz M, Altintas D, Guneser Kendirli S. Bronchiectasis: still a problem. Pediatr Pulmonol 2001; 32: 173-8.
5. Pasteur M, Helliwell S, Houghton S, Webb S, Foweraker J, Coulden R, et al. An investigation into causative factors in patients with bronchiectasis. Am J Respir Crit Care Med 2000; 162: 1277-84.
6. Lejarraga H, Krupitzky S, Heinrich J. Normas de crecimiento y desarrollo. Normas de diagnóstico y tratamiento. Buenos Aires: Sociedad Argentina de Pediatría, 1997.
7. O´Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNASe I. Chest 1998; 113: 1329-34.
8. Woodring JH. Improved plain film criteria for the diagnosis of bronchiectasis. Kansas Med Assoc J 1994; 92: 8-13
9. Polgar G, Promadhat V. Pulmonar function testing in children: techniques and standars. Philadelphia: WB Saunders, 1971.
10. American Thoracic Society. 1995. ATS statement: standardization of spirometry, 1994 update. Am J Resp Crit Care Med 152: 1107-36.
11. Hanrahan JP, Tager IB, Castile RG, Segal MR, Weiss ST, Speizer FE. Pulmonary function measures in healthy Infants. Variability and size correction. Am Rev Respir Dis 1990; 141 (suppl 1): 1127-35.
12. Chertkoff L, Visich AA. El aporte de la biología molecular al estudio de la fibrosis quística en la Argentina. Medicina Infantil 2001; 8(2): 113-9.
13. WHO Scientific Group. Primary immunodeficiency diseases. Clin Exp Immunol 1995; 99: 1-24.
14. Wilson J, Decker AM. The Surgical management of childhood bronchiectasis . Ann Surg 1982; 195: 354-63.
15. Fraser R, Paré JA. Diagnosis of Diseases of the Chest. 2nd ed. Philadelphia: Saunders, 1979.
16. Segal E, Grenoville M, Macri C, Fernández A. Consenso Argentino de Fibrosis Quística. Arch Arg Pediatr 1999; 97(3): 188-224.
17. Field CE. Bronchiectasis Third report on a follow-up study of medical and surgical cases from childhood. Arch Dis Child 1969; 44: 551-9.
18. Clark NS. Bronchiectasis in childhood. Br Med J 1963; 1: 80-7.
19. Strang C. The fate of children with bronchiectasis. Ann Intern Med 1956; 44: 630-9.
20. Glauser EM, Cook CD, Harris GBC. Bronchiectasis. A review of 187 cases with follow-up pulmonary function studies in 58. Acta Paediatr Scand (Suppl) 1966; 65: 1-10.
21. Naidich DP, McCauley DI, Khouri NF, Stitik FP, Siegelman SS. Computed tomography of bronchiectasis. Comput Assist Tomogr 1982; 6: 437-44.
22. Joharjy I, Bashi S, Abdullah A. Value of medium thickness CT in the diagnosis of bronchiectasis. Am J Roentgenol1987; 149: 1133-44.
23. Andersen DH. Cystic Fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child 1938; 56: 344–9.
24. Quinton P. Missing Cl conductance in cystic fibrosis. Am J Physiol 1986; 251: 649-53.
25. Knowles M, Stutts J, Spock A, Fischer N, Gatzy JT. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 1983; 221: 1067-70.
26. Knowles M, Gatzy J, Boucher C. Increased biolectric potential differences across respiratory epithelial in cystic fibrosis. N Engl J Med 1981; 305: 1489-94.
27. Tsui L, Buchwald M, Barker D, Braman JC, Knowlton R. Cystic fibrosis locus defined by genetically linked polymorphic DNA marker. Science 1985; 230: 1054-61.
28. Rommens J, Iannuzzi M, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of cystic fibrosis gene: chromosome walking and jumping. Science. 1989; 245: 1059-65.
29. Riordan J, Rommens J, Kerem B, Alon N, Rozmahel Z, Grzelczak J, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066-72.
30. Kerem B, Rommens J, Buchanan J, Markiewicz T, Cox A, Chakravarti M, et al. Identification of the cystic fibrosis gene:genetic analysis. Science 1989; 245: 1073-8.
31. Smith J, Travis S, Grrenberg P, Welsh D. Cystic Fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996; 85: 229-34.
32. Goldman M, Anderson M, Stolzenberg E, Kari P, Zasloff M, Wilson J. Human B- Defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in Cystic Fibrosis. Cell 1997; 88: 553-8.
33. Smith CI, Ochs HA, Puck JM. Primary Inmunodeficiency Diseases. Molecularand genetic Aproch. New York: Oxford University Press, 1999.
34. Cunningham-Randles CH, Bodian C. Commun Variable Immunodeficiency: Clinical and Immunological features of 248 Patients. Clin Immunol 1999; 92(1): 34-41.
35. Sweinberg SK, Wodell RA, Grodofsky MP, Greene JM, Conley ME. Retrospective analysis of the incidence of pulmonary disease in hipogammaglobulinemia. J Allergy Clin Immunol. 1991; 88: 96-103.
36. Buckley RH, Schiff RI. The use of intravenous immune globulin in immunodeficiency disease. N Engl J Med. 1991; 325 (2): 110-7.
37. Quartier P, Debré M, de Blic J, de Sauverzac R, Sayegh N, Jabado N, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agamma globulinemia: A retrospective survey of 31 patients. J Pediatr 1999; 134: 589-96.
Correspondencia: Dra. Ma. Catalina Pinchak
Joanico 3332/1201. Montevideo, Uruguay
E-mail: kattypi@mixmail.com

