










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.72 no.4 Montevideo dic. 2001
Enfermedad de Castleman en una adolescente
DRES. CAROLINA GARCíA 1, GUSTAVO DUFORT 2, HéCTOR PACHECO 3, CARMEN GUTIéRREZ 4, LEOPOLDO PELUFFO 5
1. Médico Residente de Clínica Pediátrica.
2. Profesor Adjunto de Clínica Pediátrica.
3. Profesor Adjunto de Clínica Quirúrgica Pediátrica.
4. Patóloga Pediátrica. Jefa del Laboratorio de Patología Pediátrica del CHPR.
5. Profesor de Clínica Pediátrica.
Recibido: 23/11/01
Aprobado: 10/12/01
Resumen
La enfermedad de Castleman es una enfermedad linfoproliferativa de las células B, de la cual existen escasas publicaciones en la edad pediátrica. Histológicamente benigna, pero que puede comportarse como maligna, su etiología aún es desconocida. Existen dos formas clínicas bien definidas: o localizada y multicéntrica o diseminada. Se describen tres tipos histológicos: hialino-vascular, plasmocítico y mixto. Puede presentarse como masa tumoral asintomática o como una enfermedad sistémica con fiebre y repercusión general.
El tratamiento óptimo no se conoce. La evolución es buena en la enfermedad de Castleman unicéntrica tras la exéresis quirúrgica, mientras que la presentación multicéntrica es de difícil manejo, con pobre respuesta a los tratamientos ensayados y mal pronóstico.
Se presenta el caso clínico de una adolescente de 14 años, con enfermedad de Castleman localizada en mediastino, que se presentó con sintomatología caracterizada por fiebre, anemia y adelgazamiento, a la cual se le practicó una resección quirúrgica completa. De los exámenes paraclínicos se destaca la presencia de una plasmocitosis en médula ósea. La respuesta al tratamiento quirúrgico fue la mejoría de los síntomas clínicos y de las alteraciones de laboratorio.
Palabras clave: HIPERPLASIA DE NÓDULO
LINFÁTICO GIGANTE
NEOPLASMAS DEL MEDIASTINO
Resumo
A doença de Castleman é uma doença linfoproliferativa das células B, da qual existem poucas publicações na idade pediátrica.
Histológicamente benigna porém, pode comportar-se como maligna, sua causa ainda é desconhecida.
Existem duas formas clínicas bem definidas: unicêntrica ou localizada e multicêntrica ou disseminada. Descrevem-se 3 tipos histológicos: hialinovascular, plasmocítico e mixto. Pode apresentar-se como massa tumoral assintomática ou como uma doença sistêmica com febre e repercussão geral.
O tratamento ótimo não se conhece. A evolução é boa na doença de Castleman localizada após cirurgia, enquanto que a doença de Castleman multicêntrica é de difícil tratamento, com pouca resposta aos tratamentos ensaiados e mal pronóstico.
Apresenta-se o caso clínico de uma adolescente de 14 anos, com uma doença de Castleman localizada no mediastino, que se apresentou com sintomatologia caracterizada por febre, anemia e emagrecimento, à qual lhe foi praticada uma ressecção cirúrgica completa. Dos exames paraclínicos, destaca-se a presença de uma plasmocitosis em medula óssea. A resposta ao tratamento cirúrgico foi a melhoria dos sintômas clínicos e das alterações de laboratório.
Palabras chave: HIPERPLASIA DO LINFONODO GIGANTE
NEOPLASIAS DO MEDIASTINO
Introducción
La enfermedad de Castleman (EC) es una enfermedad linfoproliferativa atípica, descrita por primera vez por Castleman y colaboradores en 1956. Es también conocida en la literatura como hiperplasia angiofolicular linfoide, hiperplasia de nódulo linfático gigante y, menos frecuentemente, como hamartoma linfoide, linfoma benigno o linforreticuloma folicular (1).
Histológicamente es benigna aunque puede comportarse como maligna. Se describen tres tipos histológicos: hialino-vascular (80%), plasmocítico y mixto. El hialino-vascular se caracteriza por folículos linfoides gigantes con marcada hialinización rodeado de capas concéntricas de linfocitos y proliferación vascular; y en el tipo plasmocítico se ve una acumulación masiva de células plasmáticas en el área interfolicular (1,2).
Esta enfermedad se presenta clínicamente en forma localizada (unicéntrica) o diseminada (multicéntrica).
La enfermedad de Castleman localizada (ECL) es una masa tumoral única, que puede ser asintomática o presentarse con síntomas sistémicos, de localización frecuentemente mediastinal, abdominal o periférica (3-7) y es la forma más comúnmente hallada en la edad pediátrica (1) . La enfermedad de Castleman multicéntrica (ECM) es una forma diseminada con adenopatías generalizadas, visceromegalias, manifestaciones autoinmunes, e infecciones recurrentes, en la cual se ha visto asociado el síndrome POEMS: polineuropatía, organomegalia, endocrinopatía, proteína monoclonal y rash cutáneo (8-11). El tipo histológico que se ve más frecuentemente en la enfermedad multicéntrica es el plasmocítico(1,12) .
La mayoría de los pacientes presentan síntomas sistémicos, sobre todo con el tipo plasmocítico (2,13,14) como: fiebre, sudoración nocturna, fatiga crónica, anemia y adelgazamiento (15-17).
Es una enfermedad rara en los niños. En una revisión de 83 pacientes pediátricos (13) se describe la presentación con fallo de crecimiento, retraso en el desarrollo sexual y anemia microcítica hipocrómica resistente al tratamiento con hierro.
Las alteraciones de laboratorio más comúnmente halladas son: anemia, aumento de los reactantes de fase aguda, hipoalbuminemia, hipergammaglobulinemia policlonal, citopenias autoinmunes, aumento de la interleukina-6, proteinuria y ocasionalmente insuficiencia renal (9,18-20).
Caso clínico
Adolescente de 14 años, sexo femenino, raza blanca, sin antecedentes patológicos a destacar, que es enviada desde el hospital del departamento de Artigas por desnutrición crónica severa y anemia. Presenta historia de un año de evolución de anorexia, astenia, adinamia y adelgazamiento. Retraso en la maduración sexual. Depresión y angustia importantes. Dos semanas previas al ingreso, fiebre a predominio vespertino y sudoración nocturna. Ingresa al Servicio "C" de Pediatría del Centro Hospitalario Pereira Rossell con un peso de 20 kg y una talla de 137 cm, ambos parámetros muy por debajo del percentil 3 para su edad y sexo. Del examen físico se destaca: una desnutrición severa con fundición de masas musculares, sin desarrollo de caracteres sexuales secundarios y anemia clínica.
De los exámenes complementarios cabe destacar:
- Hemograma: GB 9.000 elementos/mm3, Hb 6,6 g/dl, Hct 21.2%, MCV 72 fl, MCHC 31,2 g/dl, plaquetas 514.000 elementos/mm3, reticulocitosis 0,5%. Anisocitosis. Hipocromía moderada.
- Crasis : tiempo de protrombina 52%, fibrinógeno 947 mg/dl, KPTT 42,9 seg, tiempo de trombina 22,7 seg.
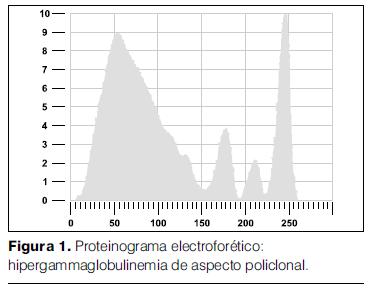
- Proteinograma electroforético: proteínas totales 13,20 g/dl, albúmina 2,22 g/dl, alfa 1: 0,48 g/dl, alfa 2: 1,05 g/dl, beta: 0,66 g/dl, gamma: 8,79 g/dl, de aspecto policlonal (figura 1).
- Dosificación de inmunoglobulinas: IgG 8651 mg/dl, IgA 334,3 mg/dl, IgM 116,4 mg/dl. VES > 140 mm/h. PCR:223,3 mg/l.
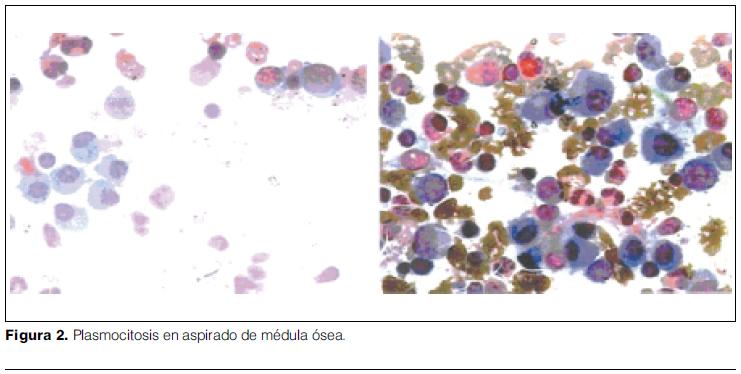
- Mielograma: médula ósea de celularidad normal. Megacariocitos presentes. Serie granulocitaria 40% en todos sus estadios de diferenciación. Serie eritroide 24%. Linfocitos 17%. Monocitos 3%. Plasmocitos 12%. Células inmaduras 4%. En suma: Plasmocitosis. Plasmocitos maduros; algunos binucleados y con cuerpos de Russell (figura 2).
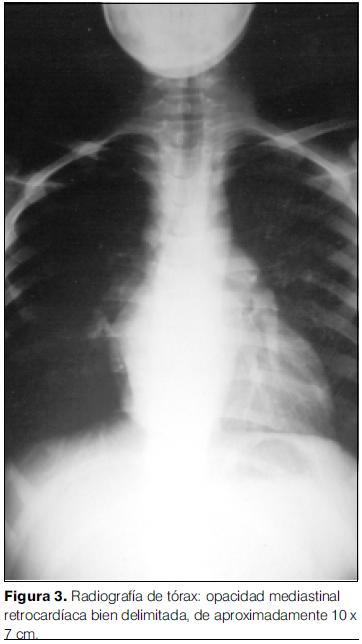
- Radiografía de tórax: opacidad mediastinal retrocardíaca densa de contornos lisos que impresiona levantar el bronquio fuente izquierdo. No hay lesiones focales pulmonares. Índice cardiotorácico aumentado para la edad (figura 3).
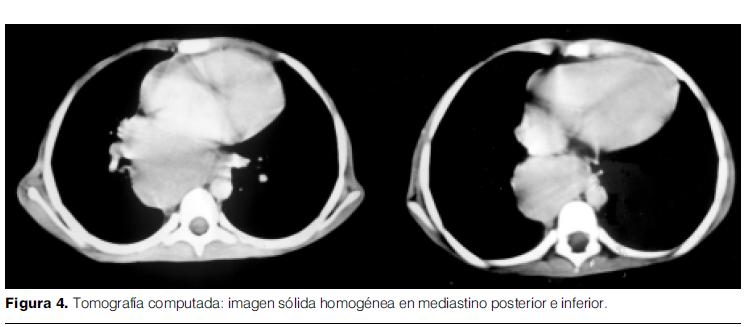
- Tomografía computada de tórax, abdomen y pelvis: imagen sólida, homogénea, de bordes nítidos, ligeramente ovalada, en mediastino posterior e inferior, de aproximadamente 8 cm. de diámetro mayor, inmediatamente por debajo de la carina, desplazando y reduciendo la luz del bronquio lobar inferior derecho, y comprimiendo aurícula derecha. Sin adenopatías mediastinales o retroperitoneales (figura 4).
A los 13 días del ingreso se realiza resección quirúrgica completa del tumor, sin incidentes, con buena evolución posterior. La anatomía patológica informa: EC de tipo plasmocítico.
Del seguimiento clínico y paraclínico de la paciente se destaca: mejoría del estado de ánimo y del apetito, con un aumento de peso de 10 kg en cuatro meses; desaparición de la fiebre y sudoración nocturna; y desarrollo de caracteres sexuales secundarios configurando un grado 2 en la escala de Tanner. Normalización de los valores de hemoglobina y de las inmunoglobulinas. VES y PCR normales. El mielograma de control muestra desaparición de la plasmocitosis. La tomografía axial computada no muestra recidiva tumoral.
Comentarios
El caso aquí presentado tiene las manifestaciones clínicas y las alteraciones de laboratorio que se describen con mayor frecuencia asociadas a la EC. El hallazgo de un tumor mediastinal determinó la conducta quirúrgica dirigida a la resección o, en su defecto, a obtener una biopsia del mismo. El tumor pudo ser resecado completamente y la evolución fue acorde a lo descrito en la literatura para las formas localizadas (21) . Un hecho a destacar en esta paciente fue el hallazgo de una plasmocitosis en médula ósea de la cual las series publicadas con mayor número de pacientes no hacen referencia. Su desaparición luego de resecado el tumor hace pensar en un origen reactivo a citoquinas segregadas por el mismo. Actualmente la paciente tiene un seguimiento de 6 meses con un espectacular retroceso de la sintomatología clínica y normalización de los exámenes de laboratorio.
La etiología de la EC es desconocida. Algunas hipótesis para su desarrollo incluyen: infecciones, autoinmunidad y fallo en la inmunorregulación (22) . La interleukina-6, una citoquina con efectos pleiotrópicos sobre el sistema inmune, la hematopoyesis y los reactantes de fase aguda, la cual se ha relacionado en la patogenia del Mieloma Múltiple en adultos, podría estar involucrada en la fisiopatología de la EC. Se ha demostrado expresión aumentada del gen codificador de la interleukina-6 en estos pacientes, con aumento de la secreción de dicha citoquina en los folículos linfoides, y mejoría de los síntomas sistémicos y de las alteraciones de laboratorio tras la administración de anticuerpos antinterleukina-6 o antirreceptor de la misma (1,9,11).
Recientemente se han detectado, mediante anticuerpos monoclonales, antígenos latentes del herpes virus humano tipo 8 (herpes virus asociado a sarcoma de Kaposi) en células plasmáticas inmaduras en algunos tipos de ECM (15,23,24).
El tratamiento óptimo no se conoce. En la ECL se han visto curaciones a largo plazo tras la exéresis completa del tumor, aunque hay descritas recidivas a los 11 años del diagnóstico, a pesar de que la resección quirúrgica había sido total (8,20,25). El riesgo de recurrencia se desconoce. Cuando el tumor no es accesible a la cirugía se ha utilizado radioterapia, resección quirúrgica parcial, corticoides o sólo observación, con el fin de evitar tratamientos muy agresivos (2,20).
La ECM es crónica. En ella se han ensayado corticoides a altas dosis, poliquimioterapia (igual a la utilizada en linfomas), ácido retinoico, interferón alfa, anticuerpos monoclonales antinterleukina-6 o antirreceptor de la misma y trasplante de médula ósea (1,9,10,15).
El pronóstico es malo en la forma diseminada, en la cual se ha descrito potencial de malignidad, sobre todo con el tipo plasmocítico (1,26), asociándose a linfoma de células B, sarcoma de Kaposi y otros, con casos rápidamente mortales y una mediana de supervivencia de 30 meses, siendo la principal causa de muerte las complicaciones infecciosas (1,8,11,20,27).
De todos modos la enfermedad en la edad pediátrica parece tener un curso más favorable que en adultos (13) .
Conclusiones
En nuestro medio no existen comunicaciones previas de EC en la edad pediátrica. El caso clínico ilustra una enfermedad poco frecuente, pero con manifestaciones clínicas y de laboratorio características que orientan a su diagnóstico. Destacamos el hallazgo de una plasmocitosis en médula ósea pues no es una de las características más frecuentes descrita en las series publicadas. Cabe destacar también la excelente respuesta al tratamiento efectuado lo cual coincide con lo observado por la mayoría de autores. Del escaso conocimiento que existe sobre la biología de esta enfermedad, es que surge la necesidad de un seguimiento a largo plazo de estos pacientes, ya sea para confirmar la curación o para despistar una posible recidiva.
Summary
Castleman`s disease (CD) is a B cell lymphoproliferative disorder, a rare disease of unknown cause. Although histologically is benign, a potential for malignancy has been described. There are two clinical types: localized (LCD) and multicentric (MCD); and three histologic variants: hyaline vascular, plasma-cell and mixed. The clinical presentation can vary from an asymptomatic localized mass to a systemic disorder with fever and constitutional symptoms.The optimal therapeutic aproach is still unknown. In the LCD complete surgical excision is usually curative, although the MCD is usually fatal with poor response to different treatment regimens.This report describes a case of CD in a fourteen-year- old girl who present a LCD in mediastinum with systemic symptoms and underwent a complete surgical excision. Laboratory values showed a remarkable plasmocytosis in the bone marrow aspirate. Both, laboratory data and clinical manifestation improve completely after surgery.
Key words: GIANT LYMPH NODE HYPERPLASIA
MEDIASTINAL NEOPLASMS
- Bibliografía
1) Herrada J, Cabanillas F. Multicentric Castleman`s disease. Clinical case reports. Am J Clin Oncol 1995; 18 ( 2): 180-3.
2) Massey G, Kornstein M, Wahl D, Huang X, McCrady C, Carchman R. Angiofollicular lymph node hyperplasia (Castleman`s disease) in an adolescent female. Clinical and inmunological findings. Cancer 1991; 68: 1365-72.
3) Salisbury JR. Castleman’s disease in childhood and adolescense: report of a case and review of literature. Pediatr Pathol 1990; 10 ( 4): 609-15.
4) Fiel-Gan MD, Voytek TM, Weiss RG, Brown RT, Joshi VV. Castleman`s disease of the left triceps in a child suspected to be a small round cell tumor of childhood. Pediatr Dev Pathol 2000; 3 ( 3): 286-9.
5) Rooney RC, Pitcher JD. Castleman’s disease in the extremity. Am J Orthop 1998; 27 ( 5): 373-4.
6) Kumar BN, Jones TJ, Skinner DW. Castleman’s disease: an unusual cause of a neck mass. ORL J Otorhinolaryngol Relat Spec 1997; 59 ( 6): 339-40.
7) Tuerlinckx D, Bodart E, Delos M, Remacle M, Ninane J. Unifocal cervical Castleman disease in two children. Eur J Pediatr 1997; 156 ( 9): 701-3.
8) Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med 1998; 128 ( 8): 657-62.
9) Beck J.T., Hsu Su-Ming, Wijdenes J. et al. Brief report: alleviation of systemic manifestation of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N Engl J Med 1994; 330 ( 9): 602-5.
10) Simko R, Nagy K, Lombay B, et al. Multicentric Castleman Disease and Systemic Lupus Erythematosus phenotype in a boy with Klinefelter Syndrome: long term disease stabilization with interferon therapy. J Pediatr Hematol Oncol 2000; 22 ( 2): 180-3.
11) Peterson BA, Frizzera G. Multicentric Castleman’s Disease. Semin Oncol 1995; 20 ( 6): 636-47.
12) Maslovsky I, Uriev L, Lugassy G. The heterogeneity of Castleman disease: report of five cases and review of the literature. Am J Med Sci 2000; 320 ( 4): 292-5.
13) Parez N, Bader-Meunier B, Roy CC, Dommergues JP. Paediatric Castleman disease: report of seven cases and review of the literature. Eur J Pediatr 1999; 158 ( 8): 631-7.
14) Spencer TD, Maier RV, Olson HH. Retroperitoneal giant lymph node hyperplasia . A case report and review of the literature. Am J Med Sci 2000; 320 ( 4): 292-5.
15) Greiner T, Armitage J.O, Gross T.G. Atypical lymphoproliferative disease. The American Society of Hematology Education Program Book. Hematology 2000; 133-146.
16) De Heer-Groen TA, Prakken AB, Bax NM, van ijken PJ. Iron resistant microcytic anaemia in a 13-year-old girl with Castleman disease. Eur J Pediatr 1996; 155 ( 12): 1015-7.
17) Powell RW, Lightsey AL, Thomas WJ, Marsh WL. Castleman’s disease in children. J Pediatr Surg 1986; 21 ( 8): 678-82.
18) Makipernaa A, Ashorn M, Arajarvi P, Hiltunen KM, Karikoski R. Castleman’s disease of the mesentery in a child: a case of seven years duration without typical X-ray findings. Med Pediatr Oncol 1997; 28 ( 5): 362-5.
19) Radaszkiewicz T, Hansmann ML, Lennert K. Monoclonality and polyclonality of plasma cells in CD of the plasma cell variant. Histopathology 1989; 14 ( 1): 11-24.
20) Bowne WB, Lewis JJ, Filippa DA et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer 1999; 85 ( 3): 706-17.
21) Grosfeld JL, Skinner MA, Rescorda FJ, et al. Mediastinal tumors in children: experience with 196 cases. Ann Surg Oncol 1994; 1: 121-127.
22) Shahidi H, Myers JL, Kuale PA. Castleman’s disease. Mayo Clin Proc 1995; 70 ( 10): 969-77.
23) Parravinci C, Corbellino M, Paulli M et al. Expresión of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman’s disease. Am J Pathol 1997; 151 ( 6): 1517-22.
24) Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 2000; 95 ( 4): 1406-12.
25) Kim JH, Jun TG, Sung SW et al. Giant lymph node hyperplasia (Castleman disease) in the chest. Ann Thorac Surg 1995; 59 ( 5): 1162-5.
26) Taylor KL, Kaschula RO. CD in children: the experience of a children’s hospital in Africa. Pediatr Pathol Lab Med 1995; 15 ( 6): 857-68.
27) Kessler E. Multicentric Giant Lymph Node Hyperplasia. A report of seven cases. Cancer 1985, 56: 2446-51.
28) O’Reilly PE, Joshi VV, Holbrook CT, Weisenburger DD. Multicentric CD in a child with prominent thymic involvement: a case report and brief review of the literature. Mod Pathol 1993; 6 ( 6): 776-80.
Correspondencia: Dra. Carolina García
Rivera 3674/102
E-mail: mugar@adinet.com.uy


{kind=link}
{kind=link}