










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Pediatría del Uruguay
On-line version ISSN 1688-1249
Arch. Pediatr. Urug. vol.72 no.3 Montevideo Sept. 2001
Síndrome de Mohr
DRAS. ADRIANA BOCCARATO 1, MARIELA LARRANDABURU 2, ÁNGELES RODRíGUEZ 3, CARMEN GUTIéRREZ 4
1. Pediatra del Servicio de Neonatología del Centro Hospitalario Pereira Rossell (CHPR).
2. Genetista clínica. Coordinadora del Estudio Colaborativo Latinoamericano de Malformaciones Congénitas (ECLAMC).
3. Patóloga pediátrica del CHPR.
4. Patóloga pediátrica. Jefe del Laboratorio de Patología Pediátrica del CHPR.
Fecha recibido: 25/5/01
Fecha aprobado: 9/7/01
Resumen
Los síndromes oro-facio-digitales representan un grupo heterogéneo de desórdenes de causa genética, caracterizados por anomalías orales, faciales y de miembros, con o sin otras malformaciones viscerales asociadas. Existen nueve tipos descritos hasta el presente. Es posible que constituyan un amplio espectro de la misma enfermedad. Los diferentes hallazgos fenotípicos se explicarían por la variable expresividad de los genes. Se presenta un caso de síndrome oro-facio-digital tipo II o síndrome de Mohr, con agenesia de cuerpo calloso.
Palabras clave: SÍNDROMES OROFACIODIGITALES
CUERPO CALLOSO- anomalías
Resumo
Os sindromes oro-facio-digitais representam um grupo heterogêneo de desordens de causa genética, caracterizados por anomalias orais, facias e de membros com ou sem outras malformações viscerais associadas. Existem nove tipos descritos até agora. Ë possível que constituam variantes diferentes da mesma doença. Os diferentes fenotipos encontrados, explicariam-se pela variável expressividade dos genes. Apresenta-se um caso de sindromes oro-facio-digitais tipo II ou sindrome de Mohr com ausência de corpo caloso.
Palabras chave: SÍNDROMES OROFACIODIGITAIS
CORPO CALOSO- anormalidades
Introducción
Los síndromes oro-facio-digitales (SOFD) fueron descritos como entidades clínicas por Papillón-Lèage y Psaume (Francia 1954).
Desde entonces se han identificado nueve tipos diferentes y se han reportado 160 casos, por lo menos, hasta el año 1995 (1). Es posible que los distintos tipos, más que tratarse de entidades separadas, constituyan un amplio espectro de la enfermedad (2), caracterizándose por defectos faciales, de la cavidad oral, anomalías de miembros y compromiso variable de otros órganos. La mayoría de los casos son familiares con patrón de herencia autosómico recesivo. Sin embargo, en el SOFD I, la herencia es ligada al X, dominante (3-5).
El síndrome de Mohr se distingue del resto fundamentalmente por la presencia de sordera de conducción, polidactilia pre y postaxial, sindactilia, con característica duplicación del hallux, raramente compromiso del sistema nervioso central y cardiopatía (6). El caso que se presenta procede de la Unidad de Perinatología del Banco de Previsión Social.
Caso clínico
Producto de cuarto embarazo de pareja no consanguínea, de 35 años la madre y 32 el padre al momento del nacimiento; tienen dos hijos vivos, sanos y un aborto espontáneo, sin estudio anátomo-patológico. Con antecedentes familiares de hermano materno portador de polidactilia (fallecido). Embarazo mal controlado, captado tardíamente y con los siguientes hallazgos ecográficos prenatales: polihidramnios, dilatación del III ventrículo, acortamiento de todos los huesos largos en forma simétrica, polidactilia (35 a 36 semanas de edad gestacional).
Parto espontáneo, en cefálica.
Recién nacido de sexo masculino, raza blanca, de término (37 semanas), peso 3.080 g (percentil 50), talla 50 cm (percentil 50), perímetro cefálico 36 cm (percentil 90).
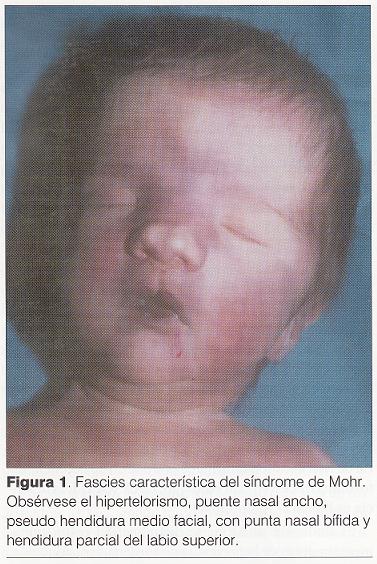
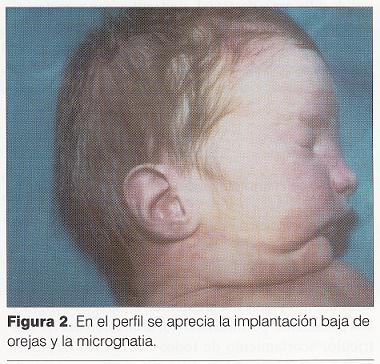
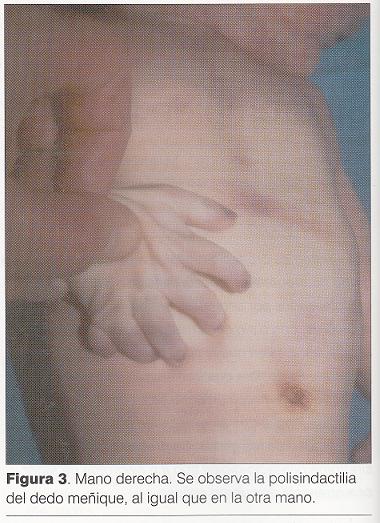
Puntuación de Apgar 3/6. Al examen físico ( y) se destaca: cráneo braquicéfalo, hipertelorismo, epicanto bilateral, raíz nasal ancha y deprimida con surco medial y punta bífida, hendidura medial del labio superior incompleta, severa micrognatia, pabellones auriculares incompletamente rotados con implantación baja, lengua de aspecto nodular con punta hendida, hendidura de paladar blando, bordes alveolares irregulares, cuello largo, tórax en quilla, hipertelorismo mamario, miembros cortos y gruesos, desviación cubital de los dedos de la mano (), polidactilia postaxil bilateral, polisindactilia de primeros y quintos dedos en ambos pies (), hipotonía marcada.
En la evolución presentó alteraciones en la succión y deglución, con mal progreso ponderal e ictericia sin conflicto que requirió fototerapia. Fue hallado muerto durante el sueño, aun internado, a los once días de vida.
Exámenes paraclínicos
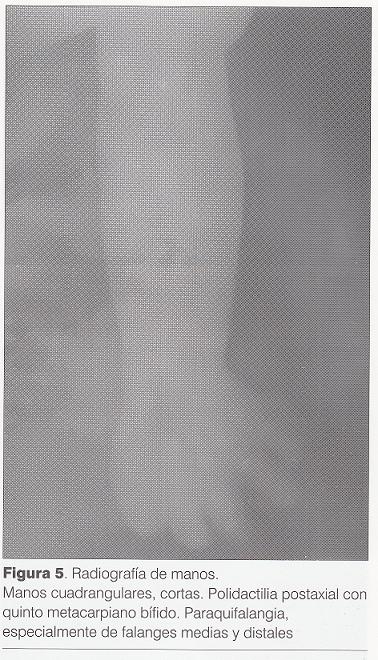
Radiografía de manos (): polidactilia postaxil con quinto metacarpiano bífido, braquifalangia de falanges medias y distales.
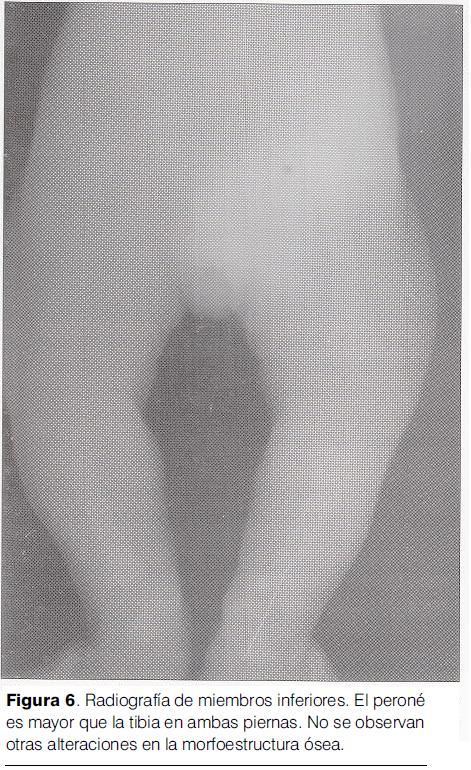
Radiografia de miembros inferiores (): peroné de mayor tamaño que la tibia, bilateralmente.
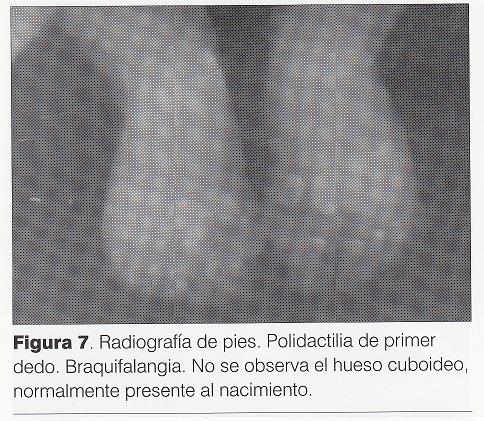
Radiografía de pies (): polidactilia de primer dedo (duplicación del hallux), braquifalangia. No se observa el cuboides, normalmente presente al nacimiento.
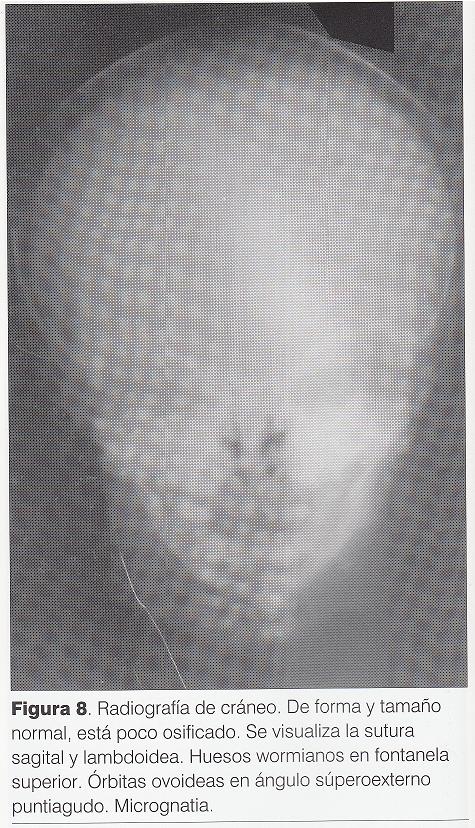
Radiografía de cráneo (): poco osificado, con huesos wormianos en fontanela posterior, órbitas ovoideas con ángulo súperoexterno puntiagudo, micrognatia.
Radiografía de tórax (): esternón de longitud mayor a lo habitual, con piezas del cuerpo más largas; costillas y vértebras normales.
Ecografía renal normal.
Ecocardiograma: comunicación interauricular pequeña, tipo ostium secundum.
Ecoencefalograma: agenesia de cuerpo calloso.
Potenciales evocados: visuales: permeabilidad de la vía visual bilateral; auditivos del tronco cerebral: hipoacusia bilateral, mayor a derecha.
Anatomía patológica: el estudio necrópsico reveló, además de las malformaciones externas ya descritas, una agenesia del cuerpo calloso y un ductus permeable. No se detectaron otras malformaciones internas.
Discusión
Las asociaciones malformativas conocidas como síndromes oro-facio-digitales representan un grupo heterogéneo de desórdenes hereditarios caracterizados por: anomalías faciales (hipertelorismo, raíz nasal ancha y deprimida, punta nasal bífida, orejas de implantación baja, micrognatia); anomalías digitales (sindactilia, polidactilia, braquidactilia, clinodactilia); anomalías de la boca (paladar hendido, labio superior hendido, frenillo lingual hipertrófico, hendidura media de lengua, hipertrofia gingival); anomalías de otros órganos. De los nueve tipos descritos a la fecha, el único de herencia dominante ligada al cromosoma X es el tipo I, que es letal en la gran mayoría de los varones afectados y que se asocia frecuentemente a malformaciones del sistema nervioso central, así como a riñones poliquísticos (3-5).
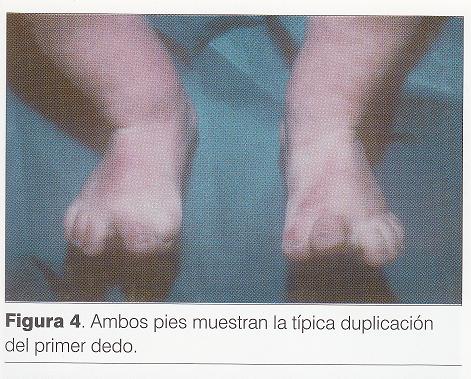
Toriello ha establecido las principales características que distinguen los tipos III al IX, de causa autosómica recesiva pero, a excepción del tipo V ( síndrome de Thurston), todos poseen anomalías en común que hacen difícil establecer el diagnóstico diferencial y el pronóstico (1). El síndrome de Mohr (SOFD tipo II), descrito por el autor en el año 1941 en gemelos varones, se caracteriza por: polidactilia pre o postaxial de manos y polisindactilia bilateral del primer dedo de pie (duplicación del hallux), punta nasal ancha, ligeramente bífida, puente nasal ancho, hendidura parcial media del labio superior, frenillo lingual hipertrófico, hipertrofia gingival, hipodontia, hendidura media de la lengua, nódulos sobre la lengua, hipoplasia de arco zigomático y cuerpo de la mandíbula (6). Existen controversias acerca de las malformaciones del sistema nervioso central en el síndrome de Mohr (7). Estos enfermos presentan baja talla y sordera de conducción. Generalmente no padecen retardo mental (8).
Conclusiones
Se ha presentado un caso de síndrome de Mohr. Se trata de un raro síndrome de causa genética. La asociación de malformaciones faciales y de miembros con o sin otras anomalías viscerales agregadas, se conocen como síndromes oro-facio-digitales.

La clasificación de los SOFD aún se basa en fenotipo clínico y en su patrón hereditario. Los casos reportados en la literatura a menudo muestran caracteres comunes a dos o tres de estos síndromes, dificultando su diagnóstico diferencial. Existe la posibilidad que los SOFD del tipo II al IX sean el resultado de una mutación genética con diferentes grados de expresividad (4). Destacamos la importancia del asesoramiento genético a la familia, ya que se trata de una enfermedad genética con riesgo de recurrencia aumentado.
Summary
The oral-facial-digital syndromes (OFD) represent an heterogeneous group of genetic disorders characterized by oral, facial and digital abnormalities. Other organ abnormalities depending on OFD type, may be included. Nine different clinical entities have been identified up to the present. The phenotypic findings could be explained as variable gene expressively and often display overlapping features between them. We report on a patient with manifestations of OFD type II or Mohr Syndrome and agenesis of the corpus callosum.
Key words: OROFACIODIGITAL SYNDROMES
CORPUS CALLOSUM- abnormalities
Bibliografía
1. Jones KL. Oral-facial-digital syndrome. In: Jones KL. Smith´s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia: Saunders, 1997: 262-3.
2. Franceschini P, Guala A, Vardeu M, Signorile F, Franceschini D, Bolgiani M. Short rib dysplasia group (with/without polydactily): Report of a patient suggesting the existence of a continuous spectrum. Am J Med Genet 1995; 59 ( 3): 359-64.
3. Leao M, Ribeiro-Silva M. Oral-facial-digital Syndrome type I in a patient with severe CNS defects. Pediatr Neurol 1995; 13 ( 3): 247-51.
4. Orstavik K, Tangsrud S, Nordshus T, Finnanger A, Hellurn C, Gjessing E. Oral-facial-digital Syndrome type I in a girl with unilateral tibial pseudarthrosis. J Med Genet 1992; 29 ( 11): 827-30.
5. Odent S, Le Marec B, Toutain A, Vigneron J, Treguier C, et al. Central nervous system malformations and early end-stage renal disease in Oral-facial-digital Syndrome type I: A review. Am J Med Genet 1998; 75 ( 4): 389-94.
6. Jones KL. Mohr Syndrome. In: Jones KL. Smith´s Recognizable patterns of Human Malformation. 5th Ed. Philadelphia: Saunders, 1997: 264-5.
7. Reardon W, Harboro M, Hall-Craggs M, Kendall B, Brett E, Baraitser M. Central nervous system malformations in Mohr´s Syndrome. J Med Genet 1989; 26: 659-63.
8. Prpic I, Cekada S, Franulovic J. Mohr Syndrome (oral-facial-digital syndrome II) - A familiar case with different phenotypic findings. Clin Genet 1995; 48 ( 6): 304-7.
Correspondencia: Dra. Adriana Boccarato.
Gauchos 4084. Montevideo.
E-mail: aboccarato@hotmail.com

