











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
“La verdad de ninguna cosa tiene vergüenza, sino de estar escondida.” Félix Lope de Vega y Carpio
Introducción
Aunque fue descubierta hace más de cien años, la mayoría de los conocimientos que contamos sobre esta patología han sido provistos en los últimos 10 años1. Su diagnóstico es un verdadero reto por su baja prevalencia, sus múltiples mutaciones y su pleomorfismo en la clínica2. La combinación de antecedentes familiares de cardiopatía y afección renal, diferentes grados de hipertrofia del ventrículo izquierdo (HVI), sumado a afecciones cutáneas, neurológicas y enfermedad renal progresiva, deben hacer plantear la posibilidad de una enfermedad de Fabry (EF)1.
En etapas tempranas el diagnóstico es planteado por pediatras, nefrólogos o dermatólogos. Dado que el compromiso cardíaco es muy frecuente con el avance de la enfermedad, en la evolución nuestro rol es igual de importante para su identificación. El cardiólogo debe plantearla ante una hipertrofia concéntrica aislada o asociada a alteraciones renales o cerebrovasculares en pacientes jóvenes. El compromiso cardiovascular es causa importante de morbilidad y mortalidad temprana en esta patología, dado por arritmias, insuficiencia cardíaca o accidentes cerebrovasculares3,4. La intención de esta revisión es describir en detalle las características clínicas, herramientas diagnósticas y terapéuticas actuales con que cuenta el cardiólogo clínico enfrentado a esta atípica patología. Con el advenimiento en 2001 de la terapia de sustitución enzimática se puede mejorar de forma radical el pronóstico si es iniciado de forma temprana, a la vez que se realiza un relevamiento diagnóstico en los familiares para evitar el futuro avance de la enfermedad2.
Epidemiología
La intensa dinámica de trabajo en policlínica de cardiología no siempre permite reflexionar sobre el diagnóstico de patologías poco frecuentes. La enfermedad de Fabry es una enfermedad rara, ampliamente infradiagnosticada. Aunque presenta una prevalencia baja, su verdadera casuística no es del todo conocida. La media desde el inicio de los síntomas hasta el diagnóstico es de 13 a 16 años en hombres y mujeres, respectivamente5.
Presenta una incidencia muy baja de 1 caso cada 40.000 a 117.000 habitantes aproximadamente en la población occidental6-8. Las variaciones amplias en las cifras se deben al subdiagnóstico, a sus distintos fenotipos con mutaciones variadas, algunas de significación clínica no aclarada, por lo que se plantea que su incidencia probablemente sea mayor.
Etiopatogenia
Es una enfermedad hereditaria de almacenamiento lisosomal, ligada al cromosoma X, causada por mutaciones en el gen GLA que codifica la enzima α-GAL A9. Esta enzima lisosomal es la encargada de degradar determinados glucoesfingolípidos, predominantemente globotriaosilceramida (Gb3)1. Estos glucoesfingolípidos no degradados se acumulan progresivamente en los lisosomas, lo que provoca cambios celulares estructurales y defectos tisulares; con el tiempo progresa a una insuficiencia orgánica, con principal implicancia del riñón, el corazón y el cerebro10.
Dado que la enfermedad está ligada al cromosoma X, un padre afectado la transmite a todas sus hijas, pero sus hijos no heredan la enfermedad porque heredan su cromosoma Y. Una madre afectada tiene un 50% de probabilidad de transmitir su cromosoma X a cualquiera de sus hijos(1). Los hombres portadores en un único cromosoma X son con mayor frecuencia afectados con las características típicas completas y de forma severa: cardiopatía, enfermedad renal, cerebrovascular, con una media de mortalidad en promedio de 56 años (la supervivencia está disminuida unos 20 años en el hombre)3.
En las mujeres heterocigotas la enfermedad exhibe una expresión pleomórfica. Rara vez presentan una afección severa al mismo grado que los hombres con afección típica. En general, presentan formas más leves e incompletas (sin afección multiorgánica) y en algunos casos tardías10. Este comportamiento puede explicarse porque tienen dos cromosomas X parentales con la inactivación aleatoria de uno de ellos; este proceso se denomina lionización. Esta inactivación al azar de uno de los cromosomas X ocurre en cada célula, en una etapa temprana del desarrollo. Las mujeres heterocigotas presentan mosaicismo, mezcla de líneas celulares con una X materna o paterna inactiva, lo que lleva a una expresión clínica de la enfermedad muy variada2.
Presentación clínica
Se identifican dos tipos clínicos de la enfermedad de Fabry de acuerdo con su presentación clínica. La forma clásica o típica y las formas atípicas1.
La forma clásica presenta compromiso multisistémico y precoz, más común en hombres homocigóticos. Las manifestaciones clínicas más frecuentes son neurológicas, dermatológicas, oculares, gastrointestinales, cardíacas y renales. Los síntomas iniciales generalmente aparecen en la niñez o adolescencia, pero frecuentemente se infradiagnostica en etapas tempranas por lo plurisintomático de la enfermedad. El diagnóstico de la enfermedad en los hombres se realiza a una edad media de 25 años y en las mujeres a los 32 años11. Las variantes atípicas presentan compromiso cardíaco o renal aislado de inicio tardío, en hombres homocigotos y mujeres heterocigotas de mediana edad7. En el trabajo de Nakao y col. alcanza una prevalencia del 3% en pacientes hombres con HVI sin clara etiología12. En una serie de pacientes varones japoneses con HVI mayor a 15 mm, su prevalencia fue del 1,1%13.
A continuación, se describen las manifestaciones de la enfermedad en diferentes órganos y sistemas:
Sistema nervioso periférico: las manifestaciones en el sistema nervioso autónomo son tempranas, comenzando en la edad pediátrica. Se presentan como dolor neuropático crónico con una incidencia alta, mayor al 80% de los pacientes14. Estos refieren acroparestesias (dolores lacerantes, urentes en manos y pies) o episodios breves de ardor intenso con crisis dolorosas en extremidades, sobre un dolor continuo profundo. Las crisis dolorosas pueden desencadenarse por temperaturas extremas, ejercicio físico o estrés. Algunos niños pueden verse impedidos de participar en actividades deportivas debido a dichas manifestaciones1,2. En adultos interfiere con ocupaciones manuales que implican ejercicio físico, tensión emocional o exposición a cambios agudos de temperatura o humedad3. También presentan una disminución de la sudoración o hipoanhidrosis que genera intolerancia al ejercicio y a las altas temperaturas4.
Sistema nervioso central: se plantea que la deficiencia de α-galactosidasa A y el depósito de Gb3 a nivel vascular lleva a una disfunción endotelial de arterias y arteriolas intracraneales. Esto genera una enfermedad vascular de pequeños y grandes vasos. La oclusión por trombosis de grandes vasos intracraneales con predominio de circulación vertebrobasilar o posterior desencadena accidentes cerebrovasculares o accidentes isquémicos transitorios, incluso en individuos jóvenes15. La edad promedio de estos eventos es de 29 años en hombres y 43 años en mujeres. Puede generar síntomas diversos como hemiparesia, mareo, vértigo, diplopía, nistagmo, disartria, cefalea, dismetría y ataxia1. Una mayor incidencia de demencia también se ha documentado relacionada con la vasculopatía cerebral. Estudios muestran que un 2-4% de la población general con accidente cerebrovascular de entre 18 y 55 años tienen enfermedad de Fabry. A su vez, se describe una mayor frecuencia de depresión y alteraciones cognitivas4.
Cutáneas: angioqueratomas. Son lesiones angioectásicas a nivel de dermis y epidermis. Se presentan como lesiones planas o poco sobreelevadas, maculopapulares de tono rojo-azulado, con vitropresión negativa (no desaparecen con la presión) y distribución corporal preferente a nivel de zona pélvica, caderas y parte superior de muslos (patrón de traje de baño). Si bien se manifiestan en etapas tempranas de la enfermedad, aumentan con su evolución16.
Sistema digestivo: se manifiestan de forma temprana en la infancia como dolores cólicos abdominales posprandiales, náuseas, vómitos y diarrea2.
Sistema auditivo: se demostró una mayor incidencia de hipoacusia progresiva (neurosensorial) y acúfenos (tinnitus) en jóvenes3.
Oculares: en general el compromiso es asintomático, presente en forma temprana y en casi la mitad de los pacientes. Bajo lámpara de hendidura se revela el signo de la córnea verticillata, generado por depósitos en el epitelio corneal de coloración grisácea- verdosa, debajo de las pupilas hacia la periferia como bigotes de gato o a rayas1,17. Puede producir también una forma de cataratas y trastornos vasculares.
Renales: una de las principales manifestaciones es la enfermedad renal por esclerosis glomerular, atrofia tubular y fibrosis intersticial (microalbuminuria, proteinuria en etapas iniciales), lo que determina un compromiso renal progresivo. Esto puede llevar a los hombres a la insuficiencia renal crónica severa y necesidad de hemodiálisis en la cuarta década de la vida18. En las mujeres pueden darse diversos grados de afectación, pero en general es de menor magnitud. Estudios en occidente estiman que entre 0,2 a 1,2% de los pacientes hombres en hemodiálisis presentan EF19. El trasplante renal mejora la morbimortalidad de estos pacientes20. Sin embargo, sin un diagnóstico y tratamiento a tiempo, los pacientes desarrollan complicaciones cardiovasculares importantes que son las que afectan más su sobrevida.
Cardiovasculares: la afectación cardíaca es muy frecuente en los pacientes con EF, tanto en la forma clásica como en la variante atípica con manifestación cardíaca. Esta es una de las principales causas de morbilidad y es la principal causa de muerte debido a insuficiencia cardíaca o arritmias ventriculares21. El compromiso cardiovascular afecta aproximadamente a un 60% de los pacientes: 69% de los hombres y 65% de las mujeres7. La prevalencia de signos y síntomas cardíacos aumenta con la edad4.
La alteración cardíaca más frecuente es la hipertrofia concéntrica, sin dilatación del VI, sin obstrucción del tracto de salida del VI y con fracción de eyección conservada22. El mecanismo etiológico de la hipertrofia ventricular se debería a la acumulación de Gb3 en los lisosomas de los miocitos, con aumento del factor trófico lisoGb3 y de esfingosina-1-fosfato que estimula la hipertrofia y la remodelación intersticial. La miocardiopatía de la EF histológicamente se caracteriza por hipertrofia y vacuolización de los miocitos y remodelación intersticial23. Esta hipertrofia aumenta con la progresión de la enfermedad. Los niveles plasmáticos de esfingosina-1-fosfato se relacionan con el grado de HVI. Los músculos papilares son prominentes en la miocardiopatía de la EF24.
En etapas avanzadas se produce fibrosis con reemplazo de miocitos, sobre todo en capas medias de la pared posterolateral basal y luego fibrosis transmural con hipoquinesia y adelgazamiento de la pared posterior del VI25. Las manifestaciones cardíacas más frecuentes incluyen disnea, ángor, palpitaciones y síncope. Clínicamente el síntoma principal es la disnea de esfuerzo por disfunción diastólica leve en etapas iniciales y severa en estadíos avanzados26.
El depósito de Gb3 a nivel de las células endoteliales y el aumento de lisoGb3 provoca aumento del músculo liso vascular, con una disminución luminal progresiva, alteración de reactividad vascular y estados protrombóticos. La cardiopatía isquémica se explicaría por vasoespasmo y enfermedad coronaria de pequeño vaso estenótico, un mecanismo diferente a la aterosclerosis19.
Uno de los signos más precoces de compromiso cardíaco en los niños es el trastorno del control autonómico de la frecuencia cardíaca, manifiesto como una disminución de su variabilidad, y bradicardia de reposo22. Debido a la infiltración del miocardio específico se producen alteraciones en la conducción, que puede progresar en etapas avanzadas a bloqueo auriculoventricular que requiere con frecuencia implante de marcapaso27. Las arritmias supraventriculares (taquicardia supraventricular, fibrilación auricular y aleteo auricular) y ventriculares (taquicardia ventricular) son frecuentes. Las arritmias ventriculares malignas vinculadas a la fibrosis miocárdica progresiva son la causa principal de morbimortalidad y mal pronóstico en los pacientes con EF28.
Métodos diagnósticos
Se basan en determinar la actividad baja o nula de la enzima α-GAL A en el plasma o los leucocitos y en identificar la mutación GLA para el gen que la codifica1. El test de la gota seca de sangre en papel de filtro que mide la actividad de la α-GAL A es un estudio con mayor especificidad en hombres dado que la actividad enzimática es baja o nula. En las mujeres heterocigotas puede estar disminuida en menor grado o ser incluso normal en plasma y reducida en órganos, por lo que un test enzimático negativo no descarta la enfermedad29.
El examen paraclínico de mayor especificidad para confirmar el diagnóstico de la EF en hombres y en mujeres es el estudio genético. Confirma el diagnóstico, determina la mutación GLA para el gen que codifica la α-GAL A y permite el estudio familiar3.
A continuación, evaluaremos los diferentes estudios a realizar para una valoración lesional completa, fundamentalmente desde el punto de vista cardiovascular:
Electrocardiograma: en etapas tempranas puede detectarse acortamiento del intervalo PR y en ocasiones trazados similares al del síndrome de Wolff-Parkinson-White. Lo más predominante es la presencia de criterios de hipertrofia ventricular, ondas R prominentes y Q anormales. Además, se presentan alteraciones del segmento ST de tipo sobrecarga ventricular e inversión de onda T anterolateral. En etapas avanzadas: bradicardia, bloqueo auriculoventricular, arritmias supraventriculares y ventriculares28.
Holter: en etapas precoces la EF genera disminución de la variabilidad autonómica de la frecuencia cardíaca. En etapas avanzadas puede presentar bradiarritmias, taquiarritmias supraventriculares y ventriculares malignas28.
Ecocardiograma: la afección miocárdica es un hallazgo predominante en más del 50% de los pacientes con EF, en general bajo la forma de HVI concéntrica30. Algunas investigaciones estiman que entre 0,5%-3% de los pacientes con HVI aislada inexplicada tienen EF31,32. Esta aumenta con la progresión de la enfermedad, pudiendo llegar a ser moderada a severa, e incluso en algunos casos simulando una miocardiopatía hipertrófica idiopática. En etapas más avanzadas la hipertrofia puede ser asimétrica, con un septum interventricular muy engrosado y menor hipertrofia o incluso afinamiento de la pared posterolateral.
En algunos casos puede observarse un aspecto binario del borde endocárdico, que no está presente en la miocardiopatía hipertrófica o miocardiopatía hipertensiva. El análisis patológico de las áreas binarias revela un endocardio y miocardio cargados de glucoesfingolípidos, separados por un espacio vacío subendocárdico. La especificidad de este signo ecocardiográfico es de 79%33.
Además, puede detectarse la presencia de músculos papilares prominentes, hipertróficos. En general no presentan obstrucción del tracto de salida del VI y la función sistólica global se encuentra preservada, solo viéndose afectada en etapas muy avanzadas34. Usando la herramienta de strain rate en ocasiones se puede demostrar una función miocárdica regional alterada incluso cuando la función global y sectorial es normal en las imágenes en 2D. Dichas anormalidades afectan principalmente la contracción longitudinal y comienzan en el segmento basal de la pared posterolateral del VI. La afección radial es tardía, en casos avanzados35.
Puede presentar diferentes grados de disfunción diastólica en la mayoría de los casos, de grado moderado a severo (patrón restrictivo)34.
Resonancia magnética cardíaca (RMC): examen de primordial importancia para evaluar la fibrosis miocárdica utilizando las imágenes de realce tardío con gadolinio, que puede guiarnos hacia el diagnóstico, evaluar el grado y estadio de afección miocárdica de la enfermedad y permitir un seguimiento. En el fenotipo más frecuente de afección cardíaca (HVI concéntrica) la fibrosis miocárdica se localiza de forma más predominante en la región posterior o inferolateral basal a nivel intramural o medial, aunque en etapas avanzadas se torna transmural33. En los casos menos frecuentes con hipertrofia ventricular asimétrica septal o hipertrofia apical, el depósito de gadolinio se concentra en el septum o el ápex, respectivamente30. La secuencia de mapeo T1 es útil al identificar acortamiento del tiempo T1 nativo (sin necesidad de utilizar gadolinio, que podría ser complejo en los pacientes con afección renal severa1.
Biomarcadores: los niveles elevados de Gb3 en plasma y/u orina pueden indicar EF en hombres, pero no tienen nivel de confiabilidad suficiente para el diagnóstico definitivo. Los niveles de Gb3 están elevados sobre todo en orina de pacientes hombres con la forma clásica y en la variante atípica con afección predominante renal, pero no se detecta en la atípica con variante cardíaca35. El biomarcador de mayor importancia es el lisoGb3 (producto de degradación del Gb3 acumulado), con elevada concentración plasmática en pacientes con EF clásica. Es útil para predecir la posible gravedad de mutaciones desconocidas en mujeres con formas atípicas de inicio tardío y test enzimáticos de alfagalactosidasa A cercanas a valor normal36.
Los péptidos natriuréticos (NT-proBNP) y las troponinas ultrasensibles se encuentran frecuentemente elevados en pacientes con mayor HVI, fibrosis y son predictores de peor evolución clínica37.
Si bien el diagnóstico muchas veces se plantea por estudios realizados en otras disciplinas, la evaluación nefrológica en la EF es primordial por su habitual afección. La microalbuminuria o albuminuria de diversa intensidad, incluso con proteinuria de rango nefrótico, puede ser detectada junto con diferentes grados de disminución del aclaramiento de creatinina, y elevación de la azoemia y creatininemia por injuria renal progresiva19.
En los casos que se realiza biopsia renal, el hallazgo en la microscopia electrónica presenta una imagen característica de la EF, los cuerpos de cebra. Estos son inclusiones en capas o multilaminares densas de lisosomas de podocitos y células epiteliales del túbulo contorneado distal38.
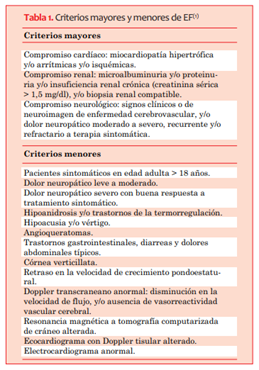
La valoración multidisciplinaria en conjunto con neurología, oftalmología y otorrinolaringología es importante, dadas las afectaciones multisistémicas mencionadas previamente. Con base en las manifestaciones clínicas más frecuentes de la EF se han establecido para su diagnóstico criterios mayores y menores1 (tabla 1).
Tratamiento
Existe un tratamiento de reemplazo enzimático (TRE) que permite un abordaje causal de la enfermedad. Es un tratamiento de muy alto costo y no exenta de complicaciones. Se dispone de dos tipos de preparados enzimáticos: agalsidasa alfa y agalsidasa beta3. En Uruguay disponemos de alfa galactosidasa A recombinante que reemplaza la enzima faltante y cataboliza los depósitos de glucoesfingolípidos.
La terapéutica sustitutiva enzimática se realiza por infusión intravenosa (dosis de 0,2 mg/kg) cada 2 semanas a lo largo de la vida. Los datos disponibles confirman la seguridad del tratamiento crónico precoz en todos los pacientes, es importante aplicarlo en etapas tempranas para enlentecer o detener la progresión lesional39. El mayor beneficio en la miocardiopatía es iniciarlo antes de que se desarrolle fibrosis miocárdica. En estos pacientes también disminuye la HVI, y la masa ventricular, a la vez que mejora la capacidad de ejercicio libre de síntomas40.
Los pacientes con afección cardiovascular se benefician de un tratamiento médico integral adecuado. Esto incluye el uso de inhibidores de la enzima convertidora de angiotensina o antagonistas del receptor de angiotensina (si no hay contraindicaciones), por sus efectos positivos en la regresión de la HVI (probados en otras formas de HVI)41. Estos fármacos tienen a su vez un efecto beneficioso a nivel renal, ya que disminuyen la proteinuria y estabilizan la función renal3. La hipotensión arterial es un efecto secundario limitante de esta terapéutica (requiere controles periódicos de presión arterial). El uso de betabloqueantes puede ser necesario para el tratamiento de taquiarritmias y prevención de arritmias ventriculares. Considerando que muchos pacientes con EF desarrollan bradicardia se debe evaluar previamente el electrocardiograma y Holter antes y durante la terapia. Como analizamos antes, los pacientes con bradicardias severas o bloqueo auriculoventricular de alto grado requieren implante de marcapaso definitivo, así como también, si se desarrollan arritmias ventriculares malignas, será necesario un cardiodesfibrilador implantable27. Debe evitarse el uso concomitante de TRE y amiodarona, dado que se han descrito interacciones. La TRE con agalsidasa alfa la financia en Uruguay el Fondo Nacional de Recursos. Debe solicitarse por formularios específicos de tratamiento y consentimiento informado, cumpliendo criterios específicos de diagnóstico y valoración lesional para ser aprobado. Posteriormente debe realizarse un seguimiento periódico con registros de las infusiones, tolerancia, formularios de seguimiento con paraclínica sanguínea y urinaria trimestral y ecocardiograma anual.
Existe actualmente una nueva terapia basada en el uso de chaperonas farmacológicas, con buenos resultados, pero no disponible en nuestro medio. Se basan en el uso de moléculas que ayudan a una proteína o enzima defectuosa a adoptar una conformación adecuada para realizar su función42.
El consejo genético es importante para el paciente y todos los miembros de su familia, dada la implicancia de la transmisión hereditaria.

