











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Introducción
Las cardiopatías congénitas del adulto (CCA) han adquirido relevancia en los últimos años debido a los avances en la cirugía cardíaca pediátrica correctiva y los procedimientos estructurales que han permitido que un número considerable de pacientes lleguen a la adultez. La población de adultos con cardiopatías congénitas crece de forma exponencial, con una tasa aproximada de 5% anual1.
La hipertensión arterial pulmonar (HAP) es una de las complicaciones más frecuentes en este grupo de pacientes. Su prevalencia es aproximadamente de 10%, y de 30% en los pacientes con defectos tipo shunt no corregidos. De estos últimos, más del 50% progresarán a fisiología de Eisenmenger, una condición patológica caracterizada por un aumento de las resistencias vasculares pulmonares e inversión de la dirección del shunt, haciéndose de derecha a izquierda o bidireccional, asociando cianosis y las complicaciones que esta conlleva2,3.
Es importante reconocer la fisiopatología del trastorno subyacente que produce en última instancia la HAP, para tratar y estratificar correctamente a estos pacientes.
Definición de HAP asociada a CCA
La HAP se caracteriza por una elevación persistente de la presión media en la arteria pulmonar, con remodelado del árbol vascular pulmonar, en ausencia de cardiopatía izquierda. La HAP es frecuente en los pacientes con CCA. En la gran mayoría de los casos resulta del aumento del flujo sanguíneo pulmonar debido a la existencia de un cortocircuito de izquierda a derecha de tamaño considerable. Este trastorno suele ser progresivo, al inicio asintomático y cuando finalmente se torna sintomático, el pronóstico empeora dramáticamente4. Existen otras etiologías más complejas que pueden generar HAP por mecanismos diversos, como se discutirá más adelante.
La terapéutica no es abundante, se dispone de un arsenal farmacológico limitado, tanto en opciones como por sus efectos adversos; así como algunas alternativas quirúrgicas o intervencionistas de rescate. El trasplante pulmonar o el combinado (corazón-pulmón) podrían constituir una opción terapéutica en pacientes seleccionados, pero limitado por la escasez de donantes.
HAP asociada a CCA: clasificación
Clínica
La clasificación clínica se basa en la dirección del flujo a través del shunt, del tamaño del defecto y de la historia de cirugías correctivas previas (figura 1)5.
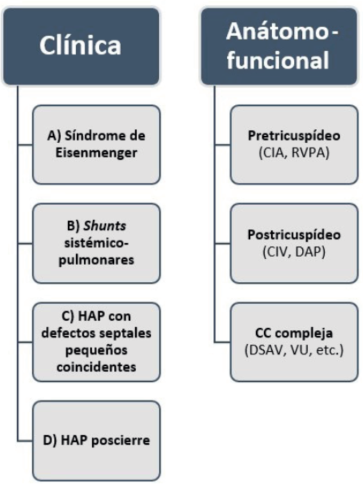
Figura 1: Clasificación clínica y anátomo-funcional de la hipertensión arterial pulmonar asociada a cardiopatías congénitas. HAP: hipertensión arterial pulmonar; CIA: comunicación interauricular; RVPA: retorno venoso pulmonar anómalo; CIV: comunicación interventricular; DAP: ductus arterioso persistente; DSAV: defectos del septum atrioventricular; VU: ventrículo único.
El grupo A consiste en el síndrome de Eisenmenger (SE), que es el extremo del espectro de las cardiopatías shunt-dependientes, en la cual la dirección del flujo a través del defecto se invierte (pasa a ser de derecha a izquierda) o se hace bidireccional, debido al aumento excesivo de las resistencias vasculares pulmonares, que determina mezcla de sangre venosa y arterial, resultando en hipoxemia y cianosis clínica de reposo.
El grupo B incluye a los pacientes con defectos moderados a grandes que permiten pasaje considerable de sangre desde la circulación sistémica a la pulmonar, resultando en hiperflujo pulmonar importante y resistencias vasculares pulmonares moderadamente elevadas. Por lo tanto, los pacientes presentan shunt de izquierda a derecha y no existe cianosis de reposo.
El grupo C consiste en pacientes con HAP y pequeños defectos septales (comunicación interauricular (CIA) < 2 cm y comunicación interventricular (CIV) < 1 cm) coincidentes. En este caso, la magnitud del shunt hacia la circulación pulmonar proveniente del corazón izquierdo no participa de la fisiopatología de la HAP. Los rasgos fisiopatológicos y clínicos de este grupo de pacientes se asemejan mucho a la HAP idiopática, por lo que se los denomina HAP con defecto septal coincidental.
El grupo D incluye los pacientes que desarrollan HAP inmediatamente o años posteriores a la corrección quirúrgica o intervencionista del defecto. Cabe destacar que estos pacientes presentan un pronóstico sombrío y que las características clínicas y fisiopatológicas son similares a la de HAP idiopática.
Anátomo-funcional
La clasificación anátomo-funcional se basa en la localización y tipo de defecto que llevó al desarrollo de HAP. En este sentido contamos con tres grupos (figura 1)5.
El grupo A son los denominados shunts pretricuspídeos, por encontrarse los defectos previos a la válvula tricúspide en el sistema circulatorio, incluye la CIA y el retorno venoso anómalo pulmonar parcial. Si no se corrigen, estos defectos producen sobrecarga de volumen en las cavidades derechas, hiperflujo pulmonar y desarrollo de HAP en etapas más avanzadas de la vida. Como el ventrículo derecho (VD) de estos pacientes se encuentra bajo regímenes de baja presión, al desarrollarse HAP es incapaz de manejar las presiones elevadas, lo cual empeora el pronóstico cuando se los compara con pacientes con HAP y shunts postricuspídeos.
El grupo B son los denominados shunts postricuspídeos, como por ejemplo la CIV y el ductus arterioso permeable (DAP). Generan sobrecarga de presión en el VD, así como hiperflujo pulmonar. Es por esto que el VD no abandona su fenotipo fetal y se encuentra “mejor preparado” para manejar presiones pulmonares elevadas, como se verá más adelante.
El grupo C lo integran las cardiopatías congénitas complejas, por ejemplo, los defectos del septum atrioventricular (o defectos tipo canal auriculoventricular), fisiología de ventrículo único, hipertensión pulmonar segmentaria, entre otros.
Fisiopatología de la HAP asociada a CCA
Los mecanismos que llevan al desarrollo de HAP vinculado a una cardiopatía congénita son multifactoriales, dentro de los cuales se incluyen vasoconstricción reactiva, proliferación de la capa media de las arteriolas pulmonares, lesiones vasculares obstructivas, inflamación y trombosis in situ6.
Con frecuencia, la HAP es secundaria a shunts de izquierda a derecha o a lesiones obstructivas en el corazón izquierdo, que determinan dos perfiles hemodinámicos diferentes.
Shunts de izquierda a derecha
Cuando la anomalía cardíaca que potencialmente puede generar HAP es una lesión tipo shunt de izquierda a derecha, el momento de la cirugía correctiva es crítico para evitar el desarrollo de enfermedad vascular pulmonar. Cuando la cirugía no ocurre a tiempo, el hiperflujo pulmonar persistente determina disfunción endotelial progresiva, vasoconstricción y remodelado vascular pulmonar (figura 2).
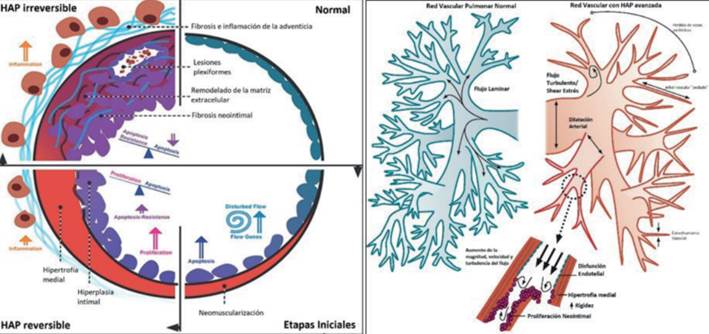
Figura 2: Izquierda. Esquema de la progresión de la lesión en la vasculatura pulmonar en la HAP, en respuesta al hiperflujo pulmonar. Las arteriolas normales tienen una única capa de células endoteliales, sin capa media prominente. El aumento del flujo genera aumento de la expresión de numerosos genes sensibles al hiperflujo que generan remodelado de la matriz extracelular, apoptosis y disfunción de las células endoteliales, así como neomuscularización. En la etapa “reversible” de la HAP por hiperflujo a través de los shunt intracardíacos, existe una hipertrofia medial y proliferación de células endoteliales, lo que genera hiperplasia intimal. En la HAP irreversible, la íntima desarrolla lesiones fibróticas, fibrosis adventicial e inflamación local. Las células endoteliales se vuelven resistentes a la apoptosis y comienzan a ocluir el lumen vascular, llevando a las características “lesiones plexiformes”. Derecha. Esquema del árbol vascular pulmonar normal y árbol vascular en HAP. La HAP se caracteriza por dilatación arterial proximal, pérdida de las ramificaciones vasculares periféricas, estrechamiento vascular y “poda” de vasos periféricos. El flujo turbulento induce los cambios esquematizados, que llevan a las lesiones neointimales, determinando obstrucción del lumen vascular y promoviendo el daño vascular. Modificado de 6.
En el caso de las lesiones tipo shunt más frecuentes, es decir, CIA, CIV y DAP, si bien el mecanismo fundamental es el hiperflujo pulmonar por cortocircuito de flujo sistémico hacia la vasculatura pulmonar, los determinantes del daño vascular pueden diferir. En el caso de los shunts pretricuspídeos, como la CIA, el grado y duración de la sobrecarga de volumen en cavidades derechas y, por lo tanto, de hiperflujo pulmonar, son los principales determinantes del daño. Sin embargo, en los shunt postricuspídeos, como la CIV y el DAP, la sobrecarga de presión y el shear estrés consecuente en la vasculatura pulmonar juegan un rol preponderante1,6.
Es importante puntualizar la diferencia de estos dos escenarios, ya que son los que con mayor frecuencia nos encontramos en la práctica clínica diaria. Los shunts pretricuspídeos son defectos de baja presión y, por lo tanto, de menor riesgo de desarrollar HAP. Cuando estas lesiones generan HAP, lo hacen a edades más avanzadas. En cuanto a los pacientes con lesiones postricuspídeas, tienen una mayor probabilidad de avanzar a SE, si no se corrige el defecto. Sin embargo, los pacientes con defectos pretricuspídeos que desarrollan SE parecen tener un peor pronóstico. En el Registro Español de Hipertensión Arterial Pulmonar, fue reportado que la mortalidad en este grupo era 2,6 veces mayor que la de los pacientes con lesiones postricuspídeas7.
La diferencia entre ambos escenarios clínicos radica en la adaptación del VD a las altas presiones. En el recién nacido, la complacencia del VD se encuentra disminuida. Esto hace que el shunt de izquierda a derecha a través de defectos como la CIA sea menor. De esta forma, la vasculatura pulmonar puede seguir su camino natural a la caída de las resistencias vasculares pulmonares (RVP) y el remodelado hacia un sistema de baja resistencia. Con el tiempo, la complacencia del VD mejora, el shunt de izquierda a derecha aumenta a través de la CIA y se produce hiperflujo pulmonar. Sin embargo, este aumento de flujo pulmonar en general no se ve acompañado de incrementos significativos de las RVP, al menos en las primeras décadas de la vida. Eventualmente, este fenómeno puede ocurrir. Si sucede y se desarrolla HAP, el VD de estos pacientes se ha adaptado a un circuito de baja resistencia (al igual que sucede con los pacientes con HAP idiopática), por lo que hay un riesgo muy alto de que este falle a medida que continúa aumentando la poscarga ventricular, conduciendo a un pronóstico sombrío. Por otro lado, en los shunt postricuspídeos, como la CIV, el VD se encuentra sometido a regímenes de alta presión, incluso antes del nacimiento. Como consecuencia, hace que este se “adapte” a un circuito de alta resistencia, otorgándole al sistema una bomba “mejor preparada” para soportar altas presiones, lo que conlleva al mejor pronóstico de estos pacientes1,8,9.
Las RVP se modifican debido al hiperflujo pulmonar mantenido y la sobrecarga de presión en el árbol vascular a través del defecto (ya sea pre o postricuspídeo), determinando una expresión alterada de los diferentes mediadores vasoactivos de la circulación pulmonar. Dentro de estos se han descrito como más importantes a las vías de la endotelina, prostaciclina y óxido nítrico, resultando en vasoconstricción persistente, fibrosis e hipertrofia de la pared vascular y degradación de la matriz extracelular vascular. Estas vías implicadas en el desarrollo de HAP son sobre las que podemos actuar farmacológicamente para enlentecer la progresión de la enfermedad (figura 3)10.
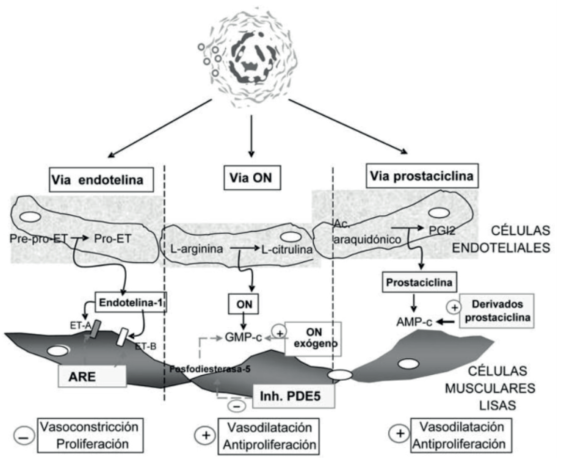
Figura 3: Mecanismos patogénicos como blancos terapéuticos en la hipertensión pulmonar. AMP-c: adenosina monofosfato cíclico; GMP-c: guanosina monofosfato cíclico; ARE: antagonista del receptor de endotelina; ET: endotelina; ETA y ETB: receptores A y B de la ET; PDE5: fosfodiesterasa tipo 5; PGI2: prostaglandina I2; ON: óxido nítrico. Tomado de 11.
Lesiones obstructivas del corazón izquierdo
En las lesiones obstructivas del corazón izquierdo, como la coartación de aorta o el complejo de Shone, la hipertensión pulmonar se produce por aumento de las presiones en las cámaras izquierdas, lo que determina un aumento de la presión arterial pulmonar media (PAPm) por aumentos de la presión de oclusión pulmonar o enclavada pulmonar. En general, estos cuadros no llevan al remodelado de la vasculatura pulmonar y, por lo tanto, al incremento de las RVP, por lo que el perfil hemodinámico es predominantemente poscapilar, como se verá más adelante12. Por esta razón, no consideramos que estos pacientes padezcan HAP.
Hipertensión pulmonar segmentaria
La hipertensión pulmonar segmentaria indica que existen segmentos del pulmón en los que existe remodelado vascular y otros con RVP normales. De esta forma, existe al menos un segmento pulmonar normotenso. Hay diversas patologías que pueden generar este cuadro, por ejemplo, atresia pulmonar, ausencia unilateral de una arteria pulmonar o arteria pulmonar de origen ductal, hemitruncus, truncus arterioso con estenosis o hipoplasia de una rama pulmonar, entre otras (figura 4).
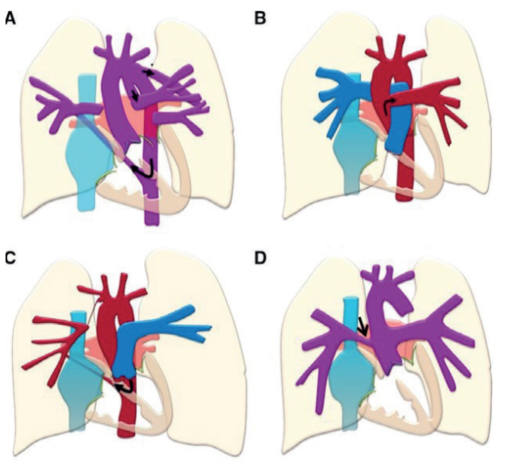
Figura 4: Ejemplos de patologías con HAP segmentaria. A) Atresia pulmonar con ramas pulmonares no confluentes, perfundidas en este caso por un ductus arterioso persistente (izquierda) y por una gran colateral aortopulmonar (derecha). B) Hemitruncus arterioso, con una arteria pulmonar izquierda que nace en el hemitruncus, y la derecha desde el tronco pulmonar en conexión directa con el ventrículo derecho. C) Ausencia unilateral de arteria pulmonar, con una arteria pulmonar izquierda única y la derecha, perfundida por una colateral aortopulmonar. D) Truncus arterioso. Modificado de 13.
La fisiopatología de la hipertensión pulmonar segmentaria es resultado del flujo pulmonar excesivo y el shear estrés sobre la pared vascular pulmonar resultante de la gran presión proveniente de grandes colaterales aortopulmonares o de orígenes anormales de las arterias pulmonares. Los segmentos pulmonares perfundidos por estos vasos anormales han mostrado en estudios anatomopatológicos hipertrofia de la capa media vascular, proliferación intimal y lesiones plexiformes. Además, se ha reportado que muchos de estos pacientes presentan un desarrollo pulmonar inadecuado, generalmente con una red vascular pequeña con capa muscular muy fina.
Cabe destacar que la perfusión pulmonar asimétrica determina un desbalance en la relación ventilación/perfusión (V/Q), que a su vez se asocia a un peor pronóstico en estos pacientes. Las colaterales que perfunden los pulmones en estas patologías llevan sangre oxigenada, lo que aumenta el espacio muerto fisiológico pulmonar.
Por otro lado, con respecto a la fisiopatología del VD en esta situación, varía según tenga flujo anterógrado hacia la circulación pulmonar en su totalidad, en parte o en ausencia de esta. Así, el estudio de estas patologías se hace sumamente complejo13.
Circulación de Fontan
La cirugía de Fontan es una cirugía paliativa que se realiza en cardiopatías complejas como la atresia pulmonar o la tetralogía de Fallot con atresia pulmonar, que se basa en una fisiología univentricular sistémica y una circulación pulmonar carente de bomba. De esta forma, se realizan conexiones desde las venas cavas superior e inferior hacia la vasculatura pulmonar (figura 5).
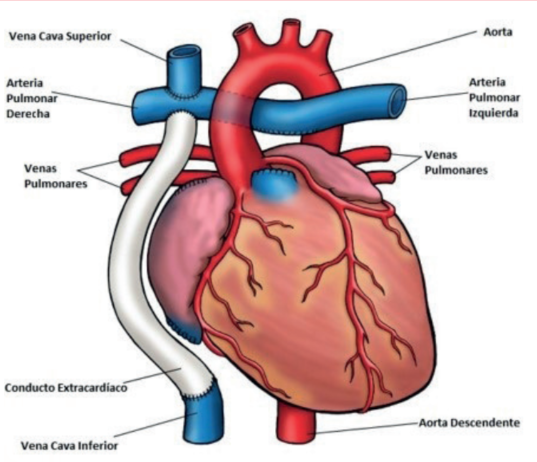
Figura 5: Esquema representativo de la circulación de Fontan. En este caso se encuentra representada la cirugía de Fontan modificada con tubo extracardíaco, la más frecuente en la actualidad. Modificado de 15.
El flujo sanguíneo pulmonar se genera de forma pasiva a través del circuito de baja resistencia. Es importante mencionar que pequeños aumentos de la PAPm o de las RVP pueden tener efectos catastróficos sobre la circulación de Fontan, determinando comorbilidades sistémicas muy importantes como falla hepática y renal. De esta manera, hay que tener en cuenta que, en los pacientes bajo este régimen circulatorio, la definición hemodinámica per se de HAP puede no cumplirse, pero sí observarse deterioro clínico por remodelado de la vasculatura pulmonar14. Ello hace de estos pacientes (entre otras características) un grupo complejo que requiere un abordaje multidisciplinario para la correcta toma de decisiones clínicas.
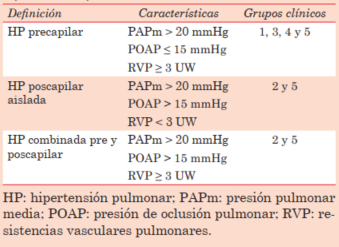
Diagnóstico hemodinámico
En el Sexto Simposio de Hipertensión Pulmonar en Niza (Francia), llevado a cabo en el año 2018, se definieron nuevos criterios hemodinámicos diagnósticos12,16. Hasta ese entonces, se consideraba diagnóstico una PAPm mayor a 25 mmHg. Hoy se considera como criterio diagnóstico una presión arterial pulmonar media PAPm de 20 mmHg en reposo, medida por cateterismo cardíaco derecho (CCD), por representar dos desvíos estándar por encima de la media poblacional: 14,0 ± 3,3 mmHg que es el valor normal de PAPm registrado en la literatura reciente. Además, ha sido recientemente reportado que los pacientes que antes se los consideraba sanos, con PAPm entre 20 y 25 mmHg, tenían un mal pronóstico comparado con los pacientes con PAPm por debajo de 20 mmHg.
Sin embargo, los valores de PAPm aislados no son suficientes para realizar el diagnóstico de HAP, ya que podrían deberse a aumentos transitorios o permanentes del gasto cardíaco, así como de la presión de oclusión pulmonar (POAP). Por esto, la POAP debe ser menor o igual a 15 mmHg. Se necesita, además, evidencia de enfermedad vascular pulmonar, por lo que debe incluir un aumento de la RVP a un valor mayor o igual a 3 unidades Wood (≥ 3 UW). Esto es necesario para realizar diagnóstico de la hipertensión pulmonar precapilar, independientemente de la etiología, que representa el perfil hemodinámico de la mayoría de los pacientes con HAP por CCA y lo encontraremos a su vez en el grupo 1 de la clasificación clínica de hipertensión pulmonar12,17.
Para los pacientes con CCA obstructivas del lado izquierdo (por ejemplo, coartación de aorta, síndrome de Shone, entre otros), el perfil hemodinámico será mayoritariamente de hipertensión pulmonar poscapilar, por lo que la característica principal será el aumento de la POAP a valores mayores a 15 mmHg12,17. Estos pacientes serán incluidos dentro del grupo 2 de la clasificación clínica de hipertensión pulmonar12.
Por otro lado, los pacientes con cardiopatías congénitas de tipo obstructivo en las arterias pulmonares (por ejemplo, estenosis congénita o hipoplasia de rama pulmonar, estenosis de ramas pulmonares poscirugía correctiva, entre otras) tendrán un perfil predominantemente precapilar y corresponden al grupo 4 según el Sexto Simposio Mundial de Hipertensión Pulmonar18.
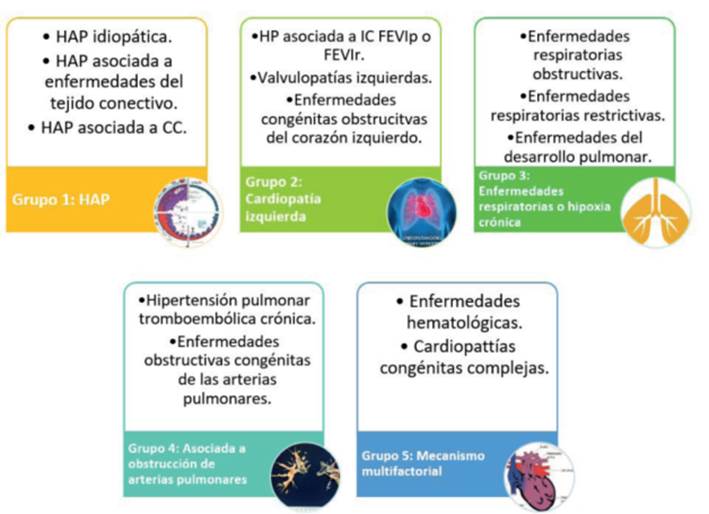
Por último, las cardiopatías congénitas complejas, como la fisiología de Fontan, o las cardiopatías asociadas a colaterales aortopulmonares, podrán tener un perfil hemodinámico de tipo precapilar o poscapilar y pertenecerán al grupo 5 dentro la clasificación clínica12,18. La clasificación hemodinámica puede verse en la (tabla 1), y las etiologías correspondientes a CCA dentro de los diferentes grupos clínicos se encuentran resumidos en la (figura 6).
Estratificación del riesgo
Es fundamental estratificar el riesgo de los pacientes que padecen HAP para planificar correctamente el tratamiento y evaluar el pronóstico a corto y largo plazo. En primera instancia, se discutirá el sistema de estratificación del riesgo utilizado para HAP que puede ser aplicado para los pacientes con CCA y luego se dedicará una sección con algunas particularidades del grupo que pueden cambiar el tratamiento propuesto.
La estratificación del riesgo en HAP fue actualizada en el último Simposio Mundial de Hipertensión Pulmonar. Este sistema incluye variables demográficas, síntomas y signos clínicos, clase funcional, capacidad de ejercicio, variables ecocardiográficas, hemodinámicas y humorales19.
En cuanto a las variables demográficas, se ha reportado que el sexo masculino es un indicador de mal pronóstico, al igual que la edad mayor a 60 años. Otra variable importante es la etiología de la HAP. La HAP asociada a enfermedades del tejido conectivo, particularmente la esclerodermia y la asociada a mutaciones genéticas, determinan un considerable peor pronóstico que las asociadas a cardiopatías congénitas20.
Cuando el paciente desarrolla síntomas y/o signos de insuficiencia cardíaca, como edema de miembros inferiores, cadencia de galope y tercer ruido, ingurgitación yugular, reflujo hepatoyugular y hepatalgia, entre otras, el pronóstico cambia radicalmente. La presencia de síncope es otra variable clínica de severidad de la enfermedad.
Otro parámetro clínico pronóstico es la clase funcional (CF) de la escala de la New York Heart Association (NYHA). La CF es una variable predictora de sobrevida muy importante. Ha sido reportado que la CF al momento del diagnóstico es una variable pronóstica de supervivencia a 5 años. Además, la CF en los sucesivos controles ha sido vinculada con ingresos hospitalarios y mortalidad19.
Es importante, además, como predictor pronóstico, la capacidad de ejercicio del paciente. Esto puede ser estudiado a través del test de marcha de 6 minutos (TM6M) o la prueba de esfuerzo cardiopulmonar, que deberían ser incorporados de forma consistente en los controles de los pacientes con HAP. Los puntos de corte para las diferentes categorías de riesgo pueden observarse en la (tabla 2)21.
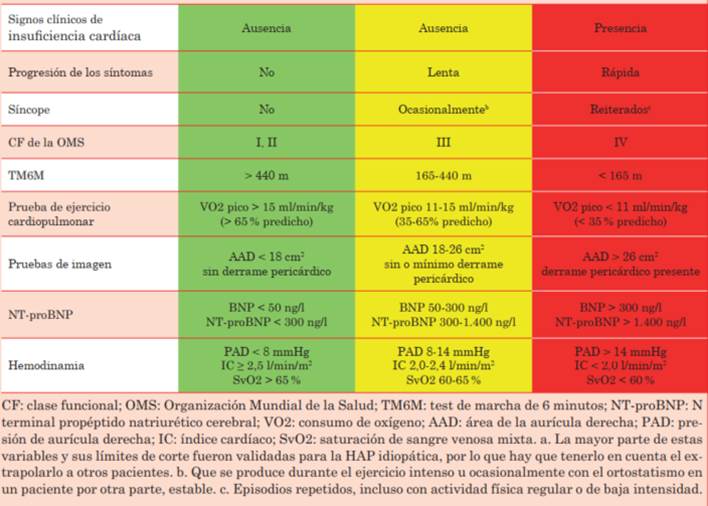
También es fundamental incluir variables imagenológicas de evaluación de la función y estructura cardíacas dentro de la estratificación del riesgo del paciente. En la ecocardiografía, es importante incluir variables de función del VD, como el desplazamiento sistólico del plano del anillo (se ha demostrado que valores por debajo de 18 mm se asocian a una disminución en la sobrevida a dos años), el acortamiento fraccional y, recientemente, el strain longitudinal global del VD. Otras variables ecocardiográficas asociadas a riesgo son el área de la aurícula derecha (AD) y la presencia de derrame pericárdico. Cuando el área de la AD supera los 18 cm2, existe un cambio en el pronóstico y, por lo tanto, un aumento del riesgo de mortalidad. La presencia de derrame pericárdico tanto al diagnóstico como en el seguimiento es un predictor importante de mortalidad y de pacientes de alto riesgo19,22.
Recientemente, se ha propuesto incluir variables obtenidas a través de resonancia magnética cardíaca. De estas variables, se ha reportado que la fracción de eyección del VD tiene un importante valor pronóstico. Incrementos en esta en el seguimiento tras tratamiento específico han demostrado asociarse a una mejoría significativa en la supervivencia a 6 años23.
Como hemos visto previamente, el CCD es fundamental para el diagnóstico de HAP y su severidad. Se ha propuesto que independientemente de su carácter invasivo, el CCD debería realizarse de 3 a 6 meses luego de cualquier cambio terapéutico del paciente, ya que otorga información valiosa y única respecto a otros estudios. Dentro de las variables hemodinámicas a evaluar, la presión de la AD, el índice cardíaco y la saturación venosa mixta han sido consideradas de mayor importancia y asociadas a peor pronóstico19.
Por último, resulta importante la medición del péptido natriurético cerebral (BNP) y/o NT-proBNP como biomarcadores, ya que se ha observado una correlación de sus valores con variables clínicas y hemodinámicas. Se ha propuesto que son útiles en el seguimiento de los pacientes y es esperable que estos desciendan luego de instaurada la terapéutica destinada a la mejoría del paciente24. La estratificación de riesgo según los parámetros discutidos se encuentra resumida en la (tabla 2).
Manejo terapéutico
Medidas generales
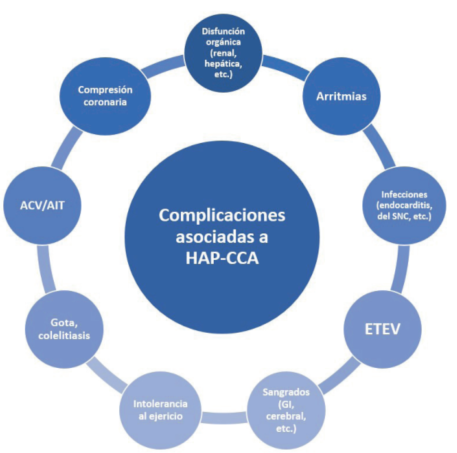
Las complicaciones de la HAP asociada a las CCA son variables y dependen del defecto subyacente, la edad del paciente, si se encuentra completa, parcial o no corregido y el grado y dirección del shunt (figura 7). Las medidas generales más importantes consisten en evitar el ejercicio físico de alta intensidad, excesivo, evitar el embarazo, apoyo psicológico y social y vacunación antigripal y antineumocócica. El oxígeno está recomendado únicamente cuando la presión parcial arterial de oxígeno es menor a 60 mmHg y en caso de SE, solo cuando genere una mejoría significativa y sostenida de la saturación de oxígeno y los síntomas3,5,25,26.
Anticoagulación
La anticoagulación en ausencia de arritmias auriculares, válvulas mecánicas o prótesis vasculares no está indicada generalmente en los pacientes con HAP asociado a CCA. La indicación en la práctica es individualizada; por ejemplo, en pacientes con grandes aneurismas de las arterias pulmonares o episodios tromboembólicos previos puede optarse por anticoagulación electiva. No existen datos de la eficacia del uso de anticoagulantes directos en pacientes con cardiopatía congénita. Existe experiencia previa en el uso de antagonistas de la vitamina K como la warfarina en estos escenarios clínicos. En el SE no está indicada la anticoagulación de rutina. Debe considerarse en aquellos pacientes con arritmias auriculares o episodios embólicos, con bajo riesgo de sangrado, ya que los pacientes cianóticos crónicos tienen mayor riesgo hemorrágico de por sí, por lo que la indicación debe considerarse caso a caso3,27.
Manejo farmacológico
Como fue explicado previamente, hay tres vías fisiopatológicas principales implicadas en el desarrollo de HAP, sobre las cuales podemos actuar a través de fármacos específicos: la vía de la endotelina (ET), las prostaciclinas y el óxido nítrico (NO)2.
Vía de la endotelina
Los antagonistas de los receptores de la endotelina (ARE) actúan bloqueando principalmente los receptores ET-A, responsables de generar un aumento del calcio intracelular que desencadena una importante vasoconstricción. Además, la activación de estos receptores lleva al desarrollo de proliferación, fibrosis e inflamación de la pared vascular pulmonar. Los ARE disponibles para el tratamiento de estos pacientes incluyen el ambrisentán (selectivo de los receptores ET-A), el bosentán y el macitentán (ambos antagonistas de receptores ET-A y ET-B)2.
Con respecto al tratamiento de pacientes con HAP y CCA con ARE, existen dos grandes ensayos clínicos randomizados que evaluaron el efecto de los ARE sobre pacientes con cardiopatías congénitas (principalmente aquellos con fisiología de Eisenmenger), que fueron los estudios BREATHE5 y MAESTRO.
El estudio BREATHE5 evaluó el efecto del bosentán sobre 54 pacientes con SE en CF de la NYHA III. El tratamiento con el ARE incrementó de forma significativa la capacidad funcional y disminuyó las RVP. Se observó que los pacientes presentaron un perfil de seguridad y respondieron de forma similar independientemente de la complejidad de la cardiopatía de base. Tampoco hubo diferencias en la respuesta según la localización del defecto septal (fuera auricular o ventricular). El tratamiento con bosentán mejoró la calidad de vida y disminuyó la mortalidad en este grupo de pacientes27.
El estudio MAESTRO fue un estudio más grande y heterogéneo en cuanto a su población, e incluyó 226 pacientes con SE en CF de la NYHA II y III. En este estudio se incluyeron, además, pacientes con síndrome de Down y cardiopatías congénitas complejas. Aproximadamente un tercio de los pacientes enrolados se encontraban bajo tratamiento con un inhibidor de la fosfodiesterasa 5 (PDE5). En este estudio, el macitentán demostró ser seguro en este grupo de pacientes y reducir significativamente las RVP y los valores de NT-proBNP en plasma28.
El ARE más comúnmente utilizado en los pacientes con CCA es el bosentán (disponible en nuestro medio). En general, este fármaco se inicia a dosis de 62,5 mg dos veces al día por cuatro semanas y se lo titula hasta la dosis objetivo de 125 mg dos veces al día. Los efectos adversos más frecuentes reportados con el tratamiento con ARE fueron la hipotensión, los edemas de miembros inferiores y la alteración de la función hepática (la cual estuvo presente en menos del 1% de los pacientes). Es esperable que hasta un 5% de pacientes presenten efectos adversos bajo tratamiento con bosentán2.
Vía del óxido nítrico
Los inhibidores de la PDE5, sildenafil y tadalafil, son los fármacos utilizados para actuar sobre esta vía. Aumentan la producción de NO al inhibir la degradación de su precursor, el GMPc, en el endotelio de la pared vascular pulmonar. El NO, a su vez, disminuye la concentración de calcio intracelular en las células musculares lisas vasculares, generando vasodilatación. Existe además un tercer fármaco, el riociguat, un estimulador soluble de la guanilato ciclasa, generando un aumento del GMPc y, por lo tanto, estimulando la producción de NO. Este fármaco puede ser utilizado también en la hipertensión pulmonar tromboembólica crónica2.
Existen varios estudios que han demostrado la eficacia del sildenafil en la HAP asociada a CCA29-32. En la mayoría de estos estudios, el tratamiento con sildenafil mejoró la clase funcional, la capacidad de ejercicio, redujo las RVP y aumentó el flujo sanguíneo pulmonar. Se ha demostrado que la combinación de sildenafil y bosentán en pacientes con HAP asociada a CCA es segura, efectiva y bien tolerada33,34.
La dosis utilizada de sildenafil varía entre 20 a 100 mg tres veces al día. En cuanto al tadalafil, la dosis recomendada es de 40 mg al día, y también ha demostrado eficacia y seguridad en el tratamiento de HAP asociada a CCA2.
Vía de las prostaciclinas
Las prostaciclinas inducen un efecto vasodilatador y antiagregante muy potente en la vasculatura pulmonar al actuar sobre los receptores IP, que activan a la adenilato ciclasa para producir AMPc. Este segundo mensajero genera relajación a nivel de las células musculares lisas pulmonares y reduce la proliferación y agregación plaquetaria in situ. Existen varias opciones comerciales de prostaciclinas sintéticas, dentro de las cuales encontramos opciones inhalatorias (iloprost), intravenosas (epoprostenol), subcutáneas (treprostinil) y orales (selexipag y beraprost)2.
Varios ensayos clínicos, inicialmente con epoprostenol en la década del 1990 y luego con fármacos más actuales en ensayos como el AIR35, ALPHABET36, los FREEDOM37-39, GRIPHON40, entre otros, han demostrado la seguridad y eficacia de este tratamiento en la HAP asociada a CCA. El iloprost inhalado, por ejemplo, demostró en pacientes con SE en CF III o IV mejorar la capacidad de ejercicio, la calidad de vida y la función del VD. La infusión continua de epoprostenol intravenoso en pacientes con HAP asociado a CCA, en los que otros tratamientos habían fracasado, demostró mejorar la capacidad de ejercicio, la saturación de oxígeno y reducir la presión pulmonar media. La terapia con estos fármacos en este grupo de pacientes ha demostrado ser efectiva y generalmente se reserva su uso para pacientes con formas severas de HAP.
La dosis de iloprost inicial recomendada (que corresponde al fármaco con el que contamos en nuestro medio) es de 2,5 mg dos veces al día por 4 semanas con una dosis objetivo de 2,5 mg cuatro a seis veces al día2.
En la (figura 8) se puede apreciar una línea de tiempo detallando los años en los que se publicaron los ensayos clínicos aleatorizados citados en la revisión.
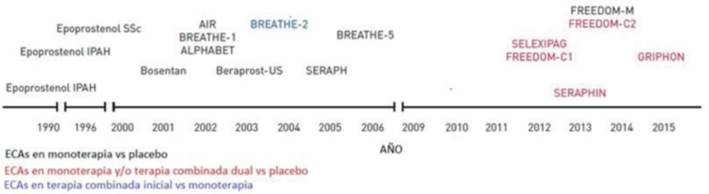
Figura 8: Línea de tiempo de los ensayos clínicos aleatorizados completados en HAP según la estrategia terapéutica. ECA: ensayo clínico aleatorizado; SSc: esclerosis sistémica; IPAH: hipertensión arterial pulmonar idiopática. Modificado de 19.
Tratamiento guiado por perfil de riesgo
Cabe destacar nuevamente que las terapias específicas para HAP no aplican a otros grupos clínicos de hipertensión pulmonar y particularmente no aplican a los grupos 2 (por cardiopatía izquierda) y 3 (vinculado a enfermedades respiratorias).
Una vez que el riesgo del paciente fue evaluado mediante los parámetros previamente discutidos, serán clasificados como de bajo, intermedio y alto riesgo19.
Los pacientes de bajo riesgo pueden ser manejados con una sola droga, o con combinación inicial de dos drogas específicas. Existe evidencia sólida de la utilidad de los ARE en el SE, mejorando sustancialmente la distancia recorrida en el TM6M, las RVP en el CCD y la CF. De esta forma en general, de optar por una terapia conservadora con un fármaco, en los pacientes con HAP asociada a CCA puede considerarse el uso de ARE y, alternativamente, inhibidores de la PDE5. De optarse por terapia combinada inicial, se recomienda utilizar un ARE asociado a un inhibidor de la PDE52,19,26.
En los pacientes clasificados como de alto riesgo, se debe iniciar triple terapia de forma inicial con un ARE, un inhibidor de la PDE5 y un prostanoide, de preferencia por vía subcutánea o intravenosa, ya que se ha reportado que los mayores beneficios de la triple terapia se alcanzan cuando se inician de forma precoz19,21.
Cuando el tratamiento inicial resulta en un paciente con perfil de bajo riesgo a los 3 a 6 meses, la terapia debe ser continuada como se planteó inicialmente y hacerse el seguimiento de forma periódica5.
En caso de que la terapia inicial resulte en un paciente con perfil de riesgo intermedio, se debe escalar a triple terapia (si la terapia inicial fue doble) o a doble terapia si se había optado por tratamiento con un solo fármaco. El algoritmo de tratamiento se encuentra resumido en la (figura 9).
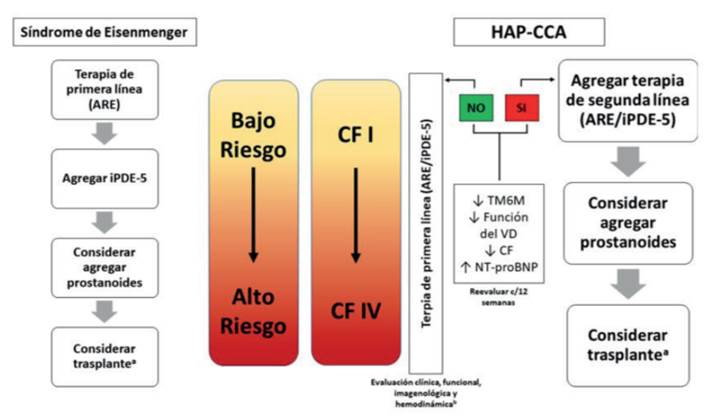
Figura 9: Algoritmo de tratamiento farmacológico específico propuesto. a. Trasplante cardíaco o cardiopulmonar. b. Evaluación cada 3 a 6 meses. Modificado de (5.
Siempre que un paciente se encuentra con triple terapia, debe ser considerado y referido a un centro de trasplante pulmonar y debe priorizarse a aquellos en los que a pesar de terapia máxima se encuentren aún en riesgo intermedio o alto.
Las medidas paliativas, como la atrioseptostomía debe resguardarse para los pacientes que a pesar de máxima terapia (tres fármacos incluyendo un prostanoide i/v o s/c), permanezcan en perfil de riesgo alto1.
Situaciones especiales
Síndrome de Eisenmenger
El SE representa el extremo más severo de la HAP asociado a CCA, donde el shunt a través de un defecto cardíaco congénito se invierte y pasa a ser de derecha a izquierda (por ejemplo, a través de una CIA, CIV o DAP), o en cardiopatías congénitas complejas con grandes colaterales aortopulmonares, provocando hipoxemia arterial y cianosis en el examen físico. Esto es consecuencia, generalmente, de la remodelación vascular pulmonar que determina aumento excesivo de las resistencias vasculares pulmonares y disminución de la capacitancia pulmonar que llevan a la inversión del flujo1.
Los avances en la cardiología pediátrica y la detección precoz de las cardiopatías congénitas han hecho que el número de pacientes que llegan a esta etapa se redujera. Sin embargo, de encontrarse el paciente en esta situación, es fundamental el diagnóstico precoz y la intervención oportuna, para evitar complicaciones asociadas a esta.
La cianosis crónica determina complicaciones multiorgánicas con un aumento sustancial de la morbilidad y hospitalizaciones, afectando negativamente la calidad de vida. La eritrocitosis secundaria se produce en respuesta a la hipoxemia crónica, que genera déficit de hierro e hiperviscosidad sanguínea. Dado que estos pacientes muchas veces asocian trombocitopenia y leucopenia, se encuentran susceptibles tanto a complicaciones trombóticas, hemorrágicas e infecciosas.
Dentro de las causas de muerte más frecuentes se encuentran la falla cardíaca, infecciones, muerte súbita y, en menor medida, causas trombóticas o complicaciones hemorrágicas. No obstante, a pesar de la gravedad de la enfermedad, los pacientes con SE suelen vivir más que los pacientes con otras formas de HAP severa. La presencia de un shunt de derecha a izquierda permite la “descarga de presión” hacia el corazón izquierdo, y mantiene el gasto cardíaco a expensas de desaturación arterial y cianosis.
Los scores de riesgo utilizados para la HAP idiopática pueden utilizarse para el SE. Cabe destacar que es fundamental estudiar la presencia de déficit de hierro, anemia y otros desórdenes hematológicos en estos pacientes.
Con respecto al tratamiento de estos pacientes, se deben considerar medidas generales y tratamiento específico. En cuanto a las medidas generales, es de destacar que los IECA y ARAII no mejoran la sobrevida en el SE, por lo que deben ser utilizados con precaución. Existe evidencia de que la digoxina aumenta la mortalidad y que los betabloqueantes podrían tener un impacto positivo en la sobrevida. El oxígeno suplementario, en ausencia de enfermedad pulmonar, no mejora el pronóstico ni la calidad de vida. Las arritmias son extremadamente mal toleradas, por lo que deben ser diagnosticadas y tratadas oportunamente. Se prefiere la cardioversión por sobre la terapia antiarrítmica de largo plazo para evitar el efecto proarrítmico de las drogas. Se debe considerar la ablación por radiofrecuencia de las arritmias supraventriculares. En caso de necesidad de dispositivos como marcapasos o cardiodesfibriladores, se deben preferir los epicárdicos y los que no tienen electrodos intravasculares, respectivamente, para minimizar los eventos trombóticos. La anticoagulación de rutina (con antagonistas de la vitamina K, por la ausencia de evidencia con la utilización de anticoagulantes directos en el SE) no ha demostrado aumentar la sobrevida, por lo que debe ser considerada si existe otra indicación para esta. Debe realizarse profilaxis de endocarditis infecciosa antes de procedimientos dentales. El embarazo se encuentra contraindicado en las pacientes portadoras de SE41.
En lo que respecta a la terapia farmacológica específica, como se mencionó previamente, debe iniciarse la terapia de pacientes sintomáticos con SE con ARE (preferentemente bosentán, por ser el utilizado en los trials que sustentan la evidencia), seguidos de inhibidores de la PDE5 como fármacos de segunda línea. De mantenerse sintomáticos a pesar de la terapia combinada, debe considerarse la adición de prostanoides i/v o s/c; por lo que el manejo farmacológico con los vasodilatadores pulmonares resulta similar a otras formas de HAP (figura 9)1.
Pacientes embarazadas con HAP y CCA
La HAP asociada a CCA acarrea una alta mortalidad maternofetal, por lo que es fundamental identificar esta situación temprano en el embarazo para intervenir con el fin de disminuir al máximo posible las complicaciones asociadas y planificar el embarazo de forma conjunta entre cardiólogos y obstetras. Sin embargo, es aún más importante brindar un consejo preconcepcional contundente, haciendo énfasis en el alto riesgo asociado a esta patología para las mujeres embarazadas (con mortalidad materna en la HAP de 16 a 30%)2.
La mortalidad materna está determinada principalmente por causas como crisis de HAP, tromboembolismo pulmonar y falla cardíaca derecha. Esto puede ocurrir incluso en pacientes con escasos síntomas previos al embarazo.
Las pacientes embarazadas con HAP asociada a CCA deben ser estratificadas según riesgo al igual que pacientes no embarazadas. La terapia combinada de manera inicial puede resultar beneficiosa, tomando en cuenta que los ERA deben ser discontinuados durante el embarazo por su teratogenicidad. Por esto, en general se ofrecen tratamientos combinados con inhibidores de la PDE5 y prostanoides. Si la paciente se encuentra bajo terapia intravenosa o subcutánea, esta debe continuarse durante el embarazo.
En cuanto al plan de parto, debe ser individualizado según el riesgo de la paciente. Debe tomarse en cuenta que siempre se prefiere la anestesia regional que la general, y que debe optimizarse previo a este lo máximo posible la función del VD, ya que es uno de los determinantes más importantes en el parto y posparto42.
Comentarios finales
La HAP asociada a CCA implica un amplio espectro de patologías muy diversas, que deben ser estudiadas y consideradas individualmente para seleccionar la terapéutica adecuada a cada paciente. A pesar de los avances en la farmacología disponible para el tratamiento de estos pacientes, la HAP continúa siendo una patología de pronóstico adverso, cuyo diagnóstico precoz y tratamiento intensivo y oportuno resultan fundamentales para modificar el curso de estos pacientes.

