











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La amiloidosis cardíaca (AC) es una cardiomiopatía (CM) restrictiva causada por el depósito extracelular de un material proteico fibrilar patológico insoluble (amiloide). Estas proteínas poseen una estructura inestable que provoca anomalías en su plegamiento, facilitando su agregación y depósito en los tejidos1. Más de 30 proteínas pueden formar fibrillas amiloides in vivo por errores de plegamiento, y en consecuencia se ha acordado basar la clasificación de la AC en su proteína precursora. La AC es causada fundamentalmente por dos tipos de proteínas, que confieren a la enfermedad diferente expresión clínica y pronóstico: a) por cadenas livianas de inmunoglobulina monoclonal (amiloidosis tipo AL) debido a una proliferación anormal de células plasmáticas, que ocurre por igual en ambos sexos; b) por transtirretina (amiloidosis tipo ATTR), una proteína sintetizada principalmente por el hígado (antes llamada prealbúmina) que circula como un tetrámero estable cumpliendo un importante papel en el transporte de la hormona tiroidea y del retinol (vitamina A) y participa en la regeneración neuronal y crecimiento axonal. La ATTR puede ser de tipo hereditaria o variante (ATTRv) como rasgo autosómico dominante causado por mutaciones patogénicas en el gen de la TTR, o bien de tipo wild type o salvaje (ATTRwt), de aparición espontánea, antes conocida como amiloidosis senil, de franca predominancia masculina2. Por razones aún no aclaradas, en la ATTRwt la estabilidad de los tetrámeros de TTR se ve afectada en ausencia de mutaciones genéticas, produciéndose monómeros y oligómeros con toxicidad tisular. A su vez, de la mutación ATTRv se han identificado un centenar de subtipos que pueden afectar el miocardio en diferente grado2,3. La proteína ATTR (ya sea ATTRv o ATTRwt) puede infiltrar otros órganos, más a menudo el sistema nervioso autónomo y periférico, aunque la afectación cardíaca constituye el principal determinante pronóstico. La mediana de supervivencia después del diagnóstico de AC en pacientes no tratados es de 2,5 años para la ATTRv y de 3,6 años para la ATTRwt4,5. La importancia de diferenciar la CM tipo ATTR (CM-ATTR) de la variedad AL (CM-AL) radica fundamentalmente en el enfoque terapéutico, en el pronóstico y en la eventual necesidad de consejo genético.
Revisión del tema
¿Por qué investigar la amiloidosis cardíaca?
En los últimos años, el interés por el diagnóstico de la CM-ATTR ha crecido como resultado de tres áreas de avance relativamente simultáneas. En primer lugar, las nuevas técnicas de imagen permiten casi siempre un diagnóstico no invasivo preciso de la CM-ATTR sin la necesidad de biopsia endomiocárdica, considerada el “estándar de oro”. En segundo lugar, varios estudios observacionales indican que la CM-ATTR es mucho más prevalente de lo que antes se creía, en especial en adultos mayores con IC y FEVI relativamente preservada (HFpEF) y en pacientes candidatos a reemplazo valvular aórtico transcatéter2,5. En tercer lugar, sobre la base del conocimiento de los mecanismos de formación del amiloide, se han desarrollado y recientemente aprobado para uso clínico algunas terapias farmacológicas especificas muy promisorias. Debido a que estas terapias para la CM-ATTR son más efectivas cuando se administran antes de la aparición de síntomas significativos de disfunción cardíaca (clase III-IV de la NYHA), la identificación temprana mediante pruebas no invasivas resulta esencial. En adición, los fármacos de uso habitual en la insuficiencia cardíaca, como los IECA, ARAII, betabloqueantes e incluso los calcioantagonistas, suelen ser muy mal tolerados por los pacientes con AC2) y, en consecuencia, el plan terapéutico debe modificarse.
Reconocimiento de la CM-ATTR
Históricamente, la CM-ATTR se ha considerado una enfermedad rara, aunque aún es difícil estimar su prevalencia real, dado que no se reconoce lo suficiente y los registros son muy limitados. Se han propuesto varias explicaciones potenciales, incluida la falsa percepción de que el diagnóstico de CM-ATTR solo se puede realizar en centros especializados mediante biopsia endomiocárdica; la atribución de los signos y síntomas del paciente al mero envejecimiento biológico; la confusión con cuadros relativamente más frecuentes como la miocardiopatía hipertrófica o la hipertensiva; y, hasta hace poco, la ausencia de tratamientos modificadores de la enfermedad, lo que restaba relevancia a la necesidad de alcanzar un diagnóstico preciso. Sin embargo, se ha demostrado depósito miocárdico de ATTR hasta en 16% de los pacientes con estenosis aórtica degenerativa y en 13-17% de los sujetos mayores masculinos hospitalizados con HFpEF5,6. Se ha reportado que casi 1 de cada 4 hombres mayores de 80 años fallecidos por cualquier causa tienen evidencia de amiloide cardíaco en la autopsia, aunque se desconoce la significación clínica de los depósitos de grado leve7. Se considera que las tasas de incidencia y prevalencia de AC están en aumento, sobre todo entre hombres mayores de 75 años; esta tendencia podría explicarse por una mayor conciencia sobre la enfermedad y, posiblemente, por un creciente uso de nuevas técnicas de imagen en la práctica clínica8.
Diagnóstico
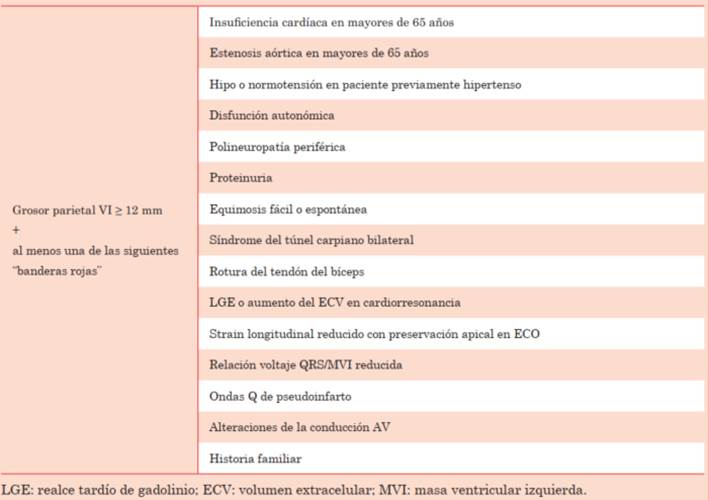
Debido a que se trata de una enfermedad infiltrativa multisistémica con depósito de amiloide en tejidos extracardíacos, los pacientes con AC presentan a menudo una constelación de signos y síntomas que conducen a sospecharla, en especial cuando se asocian a ciertos hallazgos imagenológicos y electrocardiográficos (“banderas rojas”)9. La presencia de un ventrículo izquierdo con paredes engrosadas, sumado a una o más de estas “banderas rojas”, debe orientar hacia una posible AC, como se expresa en la (tabla 1). Una vez sospechada la AC desde el punto de vista clínico, debe obtenerse un diagnóstico definitivo oportuno, ya que la evolución del paciente depende en gran medida del inicio temprano de la terapia. Los métodos auxiliarles como el ECG, el ecocardiograma (ECO), la cardiorresonancia (CR) y la medicina nuclear (MN), junto con los datos de laboratorio, permiten confirmar el diagnóstico y eventualmente diferenciar la variedad responsable.
Aporte de la medicina nuclear
A diferencia de los otros métodos, la imagen a través de la MN (centellograma cardíaco con fosfatos marcados) permite casi siempre diferenciar entre los dos tipos más comunes de amiloidosis sin necesidad de recurrir a la biopsia endomiocárdica, por lo cual hoy se considera equivalente a una biopsia molecular10.
Radiotrazadores
La imagen cardíaca con trazadores de afinidad ósea (varios de ellos de utilización habitual para centellografía ósea) fue empleada décadas atrás para el diagnóstico de infarto de miocardio, por la avidez de estos radiofármacos por los depósitos tempranos de calcio en el tejido necrótico. Aunque cayó en desuso ante la aparición de métodos más precisos. Algunos estudios ya demostraron entonces captación miocárdica en la AC, aunque su utilidad clínica fue escasamente investigada11. Más recientemente, varios investigadores confirmaron la capacidad de los difosfonatos marcados con 99mTc para unirse al amiloide tipo ATTR, lo cual condujo al renacimiento de la imagen centellográfica y su inclusión en los algoritmos diagnósticos de la AC. Tres radiotrazadores con afinidad ósea pueden ser empleados con este propósito: el 99mTc-pirofosfato (99mTc-PYP), el 99mTc-ácido difosfono1,2propanodicarboxílico (99mTc-DPD) y el 99mTc-hidroximetilendifosfonato (99mTc-HMDP). El 99mTc-metilendifosfonato (99mTc-MDP), ampliamente utilizado en la actualidad para centellografía ósea, ha mostrado menor sensibilidad, y no se considera, por tanto, el trazador de preferencia para identificar CM-ATTR12. El mecanismo de captación de los trazadores fosfatados en la AC no se encuentra totalmente dilucidado, aunque se cree vinculado a la presencia de calcio en los depósitos fibrilares13,14, en especial en la variedad ATTR. Sin embargo, si bien se ha demostrado en biopsias endomiocárdicas de CM-ATTR una mayor densidad de microcalcificaciones en comparación con la CM-AL, algunos casos de AL pueden exhibir una densidad similar a la ATTR, lo que explicaría los reportes border line positivos con 99mTc-PYP en ciertos pacientes con CM-AL15,16.
Protocolo de imágenes
Luego de la administración de 15-20 mCi de 99mTc-PYP (u otro trazador óseo apropiado) se obtienen imágenes planares del tórax en diferentes proyecciones entre 1 y 3 horas después de inyectado el radiofármaco, seguidas de un estudio tomográfico (SPECT)12,17,18. Como muchos de estos pacientes presentan bajo gasto cardíaco y/o insuficiencia renal, el aclaramiento sanguíneo del trazador puede estar enlentecido, por lo cual las imágenes tardías y el SPECT son a menudo necesarios para aumentar la especificidad del estudio, a fin de evitar la confusión con la persistencia del radiofármaco en el espacio vascular (pool sanguíneo)18-20. Algunos autores sugieren incluir el cuerpo entero en la exploración, para investigar compromiso sistémico18.
Interpretación de las imágenes
La interpretación de las imágenes con fosfonatos marcados requiere simplemente evaluar la presencia del radiotrazador en el área de proyección cardíaca y su comparación con la captación ósea fisiológica correspondiente al esternón y la parrilla costal, estableciendo una gradación visual o por métodos semicuantitativos. Perugini y col.12 propusieron el uso de una sencilla escala visual para las imágenes de 3 horas, desde grado 0 a grado 3 según la intensidad de captación en el área cardíaca y su relación con la captación ósea (figura 1). Un estudio multicéntrico internacional que incluyó 1.271 pacientes referidos por sospecha de AC demostró que un puntaje de Perugini ≥ 2 en ausencia de gammapatía monoclonal determinada en sangre y orina se asocia a una especificidad y valor predictivo positivo de 100% para AC-ATTR, con una sensibilidad del 72%17. Sin embargo, hasta 20% de los pacientes tuvieron un pico de proteína monoclonal en las pruebas de laboratorio y la especificidad se ubicó en 91% en los pacientes en que no fue posible excluir AL por ese medio. Este hecho resalta la absoluta necesidad de contar con dosificación de cadenas livianas mediante estudios de inmunofijación al tiempo del estudio centellográfico. Bokhari y col.15 desarrollaron un método de análisis semicuantitativo mediante un índice de captación corazón/tórax contralateral (C/CL) en las imágenes planares (figura 2) para distinguir la amiloidosis de tipo ATTR de la AL, con un valor de corte de 1,5 a los 60 minutos posinyección o de 1,3 a las 3 horas. El uso de esta metodología aplicando dichos valores de corte ha demostrado una sensibilidad y especificidad de 92% y 97% y de 88% y 86%, respectivamente19.
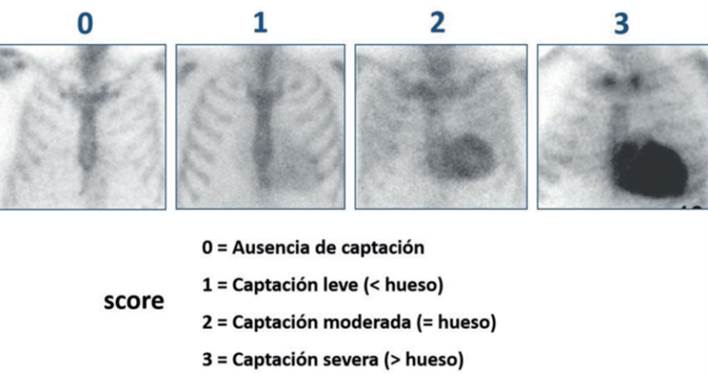
Figura 1: Escala de Perugini según la captación miocárdica relativa al hueso. En ausencia de cadenas livianas, una captación grado 2 o 3 es patognomónica de CM-ATTR.
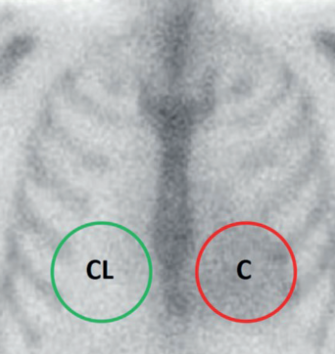
Figura 2: Índice C/CL. Se mide la actividad (cuentas) en el área cardíaca (C) y se divide por la registrada en un área en espejo sobre el hemitórax contralateral (CL). Un valor ≥ 1,5 a una hora posinyección o ≥ 1,3 a las 3 horas sugiere ATTR.
Como en todo método diagnóstico, el centellograma puede tener resultados falsos positivos y falsos negativos que deben considerarse (tabla 2). Los primeros se vinculan a algunos tipos de amiloidosis AL (AApoAI, AApoAII, ApoAIV y Aß2M), que generalmente están asociadas a compromiso renal. Otras causas de falsos positivos, como la persistencia del radiotrazador en el espacio vascular, fracturas costales, calcificaciones valvulares, infarto de miocardio reciente (menor a 4 semanas), pueden ser en general identificadas mediante SPECT o mediante técnica híbrida SPECT/CT (figura 3). También se ha descrito captación miocárdica incidental en otras condiciones como la enfermedad de Chagas, cardiotoxicidad por hidroxicloroquina, miocarditis, trauma cardíaco y cardioversión reciente11,25. En cuanto a los falsos negativos, pueden darse en algunos subtipos raros de la ATTR hereditaria como la Pne84Leu-ATTRv o la Ser97Tyr-ATTRv, que concomitan con neuropatía y enfermedad familiar y se identifican mediante pruebas genéticas. Otras causas de falsos negativos son la afectación cardíaca leve o incipiente y ciertos aspectos técnicos como la precocidad o el retraso en la adquisición de las imágenes; todo esto destaca la necesidad de un adecuado grado de experticia para la realización e interpretación del estudio9.
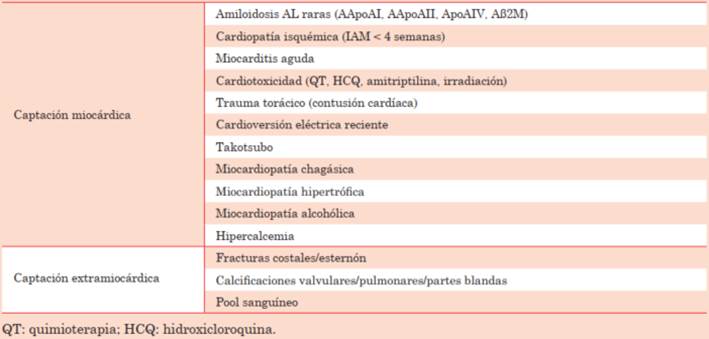
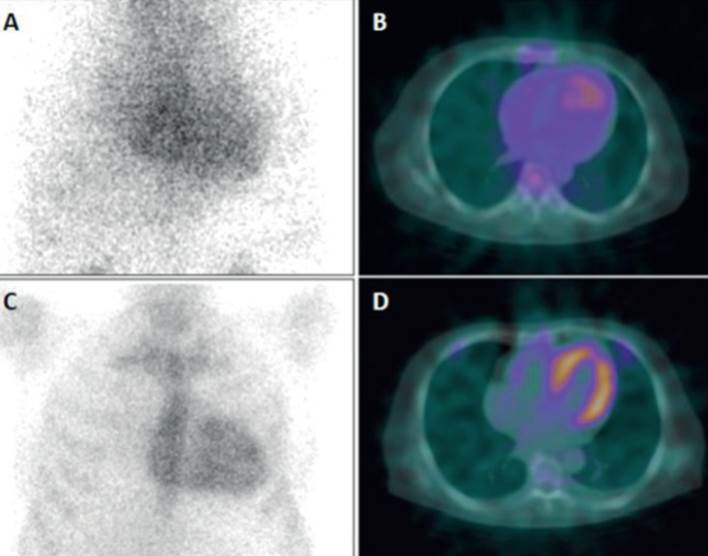
Figura 3: A) Imagen planar de tórax que muestra captación de 99mTc-PYP en el área cardíaca, en forma difusa (Perugini 1 2). B) SPECT/CT en plano transaxial confirma que la captación corresponde al pool sanguíneo y no a la pared miocárdica (negativo para ATTR). C) Captación cardíaca relativamente definida en la imagen planar (Perugini 2). D) SPECT/CT demuestra claramente la captación miocárdica (cortesía Dr. Gabriel Grossman, Porto Alegre).
Algoritmo
Dado que la gran mayoría de los casos de AC son de tipo AL o ATTR, la Sociedad Europea de Cardiología, la American Heart Association y otras sociedades científicas proponen algoritmos diagnósticos similares y relativamente sencillos dirigidos a identificar estos subtipos mediante el uso inicial de MN con 99mTc-PYP, DPD o HMDP junto con proteinograma electroforético sérico y urinario con inmunofijación y cuantificación de cadenas livianas libres en suero9,20,21. Los resultados de esta combinación de pruebas podrían conducir a los siguientes escenarios (figura 4):
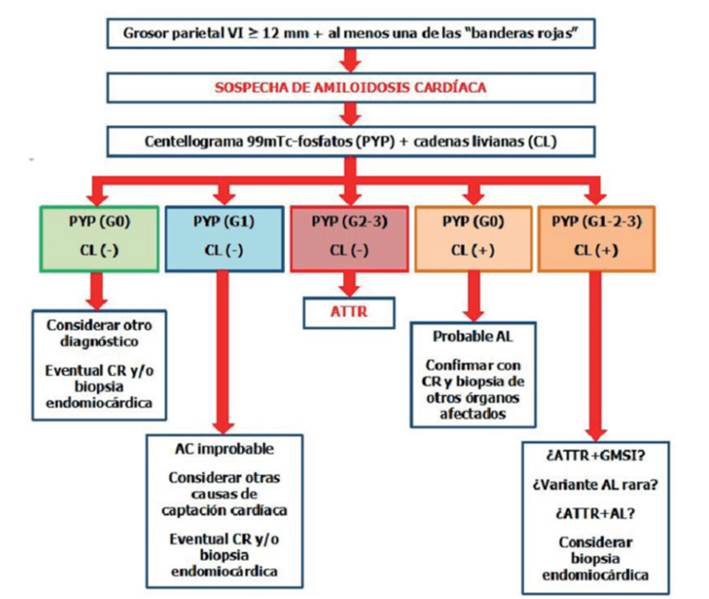
Figura 4: Algoritmo diagnóstico basado inicialmente en el uso simultáneo de centellograma con fosfatos marcados (99mTc-PYP) y dosificación de cadenas livianas (CL) que da lugar a 5 escenarios posibles. GMSI: gammapatía monoclonal de significado incierto.
1. Ausencia de captación cardíaca (grado 0) y evaluación de proteínas monoclonales negativa. Existe muy baja probabilidad de AC y debe considerarse un diagnóstico alternativo. Si persiste la sospecha, se debe considerar CR seguida de biopsia cardíaca o extracardíaca, ya que la MN podría ser negativa en algunas mutaciones raras de ATTRv.
2. Captación cardíaca grado 1 y evaluación de proteínas monoclonales negativa. Se deben descartar razones técnicas de un estudio dudoso (pool sanguíneo); si persiste la duda, considerar CR seguida de biopsia cardíaca o extracardíaca.
3. Captación cardíaca grado 2 o 3 y evaluación de proteínas monoclonales negativa. Se puede establecer el diagnóstico de CM-ATTR y eventualmente continuar con pruebas genéticas para diferenciar entre ATTRv y ATTRwt.
4. Ausencia de captación cardíaca (grado 0) y al menos una de las pruebas de proteínas monoclonales positiva. La CM-ATTR puede descartarse y debe confirmarse una probable CM-AL mediante CR. Sin embargo, si el resultado es negativo, el diagnóstico de AC es muy poco probable, mientras que si no es concluyente, se requiere biopsia extracardíaca (médula ósea u otros órganos clínicamente afectados), o cardíaca si la anterior es negativa.
5. Captación cardíaca (grado 1,2 o 3) y al menos una de las pruebas de proteínas monoclonales anormal. En tal caso es posible la concomitancia de CM-ATTR con una gammapatía monoclonal de significación indeterminada o cualquier trastorno hematológico que produzca cadenas livianas. También es posible alguna variedad de amiloidosis AL (sobre todo si la captación es grado 1 o 2), o incluso la excepcional coexistencia de AL y ATTR. En este escenario, el diagnóstico de AC requiere histología con tipificación amiloide, generalmente mediante biopsia endomiocárdica.
Valor pronóstico
En 1997 Dubrey y col.22 reportaron, en el seguimiento a un año, una sobrevida de 92% para los pacientes portadores de CM-ATTR y de solo 38% para el tipo AL. Establecieron la importancia de diferenciar ambas variedades dada su implicancia en cuanto a pronóstico y tratamiento. La literatura disponible muestra la capacidad del método centellográfico semicuantitavo (índice C/CL) para establecer el riesgo de eventos cardíacos mayores; sin embargo, no se ha podido determinar que el score visual de Perugini por sí mismo sea un predictor independiente. En un estudio multicéntrico retrospectivo de 229 pacientes, Castaño y col.19 enfatizan la importancia pronóstica del índice C/CL, ya que un valor de ≥ 1,5 estuvo asociado a peor sobrevida en un período de 5 años, mientras que un score Perugini grado 2 o 3 no lo estuvo de modo significativo, priorizando el método semicuantitativo sobre el visual. Por otro lado, Hutt y col.23 reportan que los únicos predictores independientes de mortalidad en su serie fueron el estado funcional definido por ECO, el score visual con 99mTc-PYP (grado 0 vs. grado 1, 2 o 3) y la función renal (filtración glomerular) después de ser ajustados por los niveles de NT-proBNP, edad, FEVI y presión arterial de pie (< 100 mmHg). Otros investigadores han estudiado el pronóstico desde el punto de vista de los marcadores bioquímicos, en particular las troponinas T y el NT-proBNP, estableciendo distintos estadios según el número de parámetros afectados, con sobrevidas estratificadas entre 66 y 20 meses24,25.
Conclusiones
La AC es una enfermedad subdiagnosticada, de elevada mortalidad y prevalencia creciente con el aumento de expectativa de vida de la población. Se debe considerar la AC en pacientes adultos mayores con aumento del grosor de la pared ventricular en presencia de señales de alerta (“banderas rojas”) cardíacas o extracardíacas, en especial IC con FEVI relativamente preservada. Un algoritmo basado inicialmente en el uso de la MN con compuestos fosfatados (pirofosfato u otros), junto con la evaluación de proteínas monoclonales, conduce a un diagnóstico preciso en la gran mayoría de los casos y permite la implementación temprana de medidas terapéuticas específicas.

