











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
PermalinkIntroducción
La miocardiopatía no compactada (MCNC) representa una anomalía de la morfología miocárdica frecuentemente asociada a una etiología genética, caracterizada por la estructuración del miocardio en dos capas diferenciadas: a) Una capa endocárdica gruesa de aspecto esponjoso, con una trabeculación prominente, entendiendo las trabéculas desde un punto de vista ecocardiográfico como estructuras con ecogenicidad similar a la del miocardio y que se mueven sincrónicamente con la contracción del resto del ventrículo1 y recesos profundos que comunican con la cavidad ventricular pero no con la circulación coronaria, y b) Otra capa epicárdica de aspecto denso y uniforme que suele ser más delgada que la primera2. Aunque habitualmente afecta al ventrículo izquierdo (VI), puede afectar a ambos ventrículos e incluso al ventrículo derecho (VD) de forma exclusiva.
Tiene un curso clínico altamente variable pero potencialmente mortal, ya que en algunos pacientes ocasiona disfunción sistólica e insuficiencia cardíaca, arritmias ventriculares y complicaciones tromboembólicas. Su clasificación es altamente controvertida, dado que la American Heart Association (AHA)3) la considera una miocardiopatía primaria de origen genético, mientras que la World Health Organization (WHO)4) y la European Society of Cardiology (ESC)5 la definen como una miocardiopatía inclasificada. Incluso se duda de que se trate de una miocardiopatía claramente definida y no constituya realmente la manifestación fenotípica de otras enfermedades subyacentes2,6.
Epidemiología
Realmente es desconocida, con datos muy variables según los estudios actualmente disponibles, dado que a menudo estos son retrospectivos, con sesgos de selección, diferentes criterios diagnósticos, etcétera. Entre los pacientes derivados al laboratorio de ecocardiografía se estima una frecuencia de entre el 0,014% y el 4%7, siendo la edad media en el momento del diagnóstico de 40 años en población adulta y de 7 años en niños. En estudios en población infantil con miocardiopatías primarias, corresponde al 9,2% de los casos, siendo la tercera causa más frecuente luego de la miocardiopatía dilatada y la hipertrófica8. En clínicas específicas de insuficiencia cardíaca se estiman prevalencias del 3% de todos los pacientes9. Con la mejora en las técnicas de imagen, esta patología se está diagnosticando crecientemente, de tal forma que incluso pudiera existir un problema de sobrediagnóstico. Algunos estudios apuntan a un mayor predominio del cuadro en hombres. Las regiones apicales y la pared inferolateral media del VI suelen ser las más afectadas10.
Fisiopatología
La presencia de trabeculaciones cardíacas es un fenómeno totalmente fisiológico en el desarrollo embriológico del corazón. Hay presencia de las mismas ya desde la cuarta semana de gestación, lo que permite aumentar la superficie sobre la que se va a desarrollar la masa miocárdica futura en ausencia todavía de un aporte coronario epicárdico. A partir de la octava semana ocurre un proceso de remodelado de las trabéculas y de compactación del miocardio, cuando ya existe un aporte sanguíneo coronario propio. Este proceso de compactación se inicia desde el epicardio al endocardio, desde la base del corazón al ápex y desde el septo a la pared lateral. La detención de este fenómeno originaría la persistencia de una capa de trabéculas prominentes separadas por recesos profundos en comunicación con la cavidad ventricular (lo que correspondería a la capa no compacta)11,12.
La presencia de trabeculaciones prominentes y no compactación puede ser hereditaria, pero también puede aparecer de novo o incluso adquirirse con el tiempo en situaciones fisiológicas o patológicas, bien de forma totalmente aislada o acompañando a otras miocardiopatías, anomalías congénitas13,14 (enfermedad de Ebstein, obstrucción del tracto de salida del VI, válvula aórtica bicúspide, cardiopatías cianóticas congénitas y anomalías coronarias), síndromes genéticos complejos, trastornos metabólicos, y especialmente asociada a trastornos neuromusculares (hasta en un 21% de los casos)15, como distrofia de Becker, miopatía metabólica, distrofia miotónica tipo 1, enfermedad de Pompe, neuropatía óptica de Leber, ataxia de Friedrich y enfermedad de Charcot-Marie-Tooth. Por este motivo, se recomienda despistaje neurológico en caso de MCNC y viceversa.
Mutaciones en numerosos genes16 se han asociado al desarrollo del cuadro. Estos genes están principalmente involucrados en codificar proteínas sarcoméricas, del citoesqueleto, mitocondriales o de la membrana nuclear tales como la tafacina, beta DTNA, LDB3, lamina A/C, SCN5A, MYH7 o MYBPC36. Si bien la genética de la enfermedad está siendo aún estudiada, se ha comprobado que muchos casos hereditarios se asocian a mutaciones en los mismos genes que causan otros tipos de miocardiopatías (genes que codifican proteínas sarcoméricas relacionadas con la miocardiopatía hipertrófica, restrictiva y dilatada), superponiéndose a su expresión fenotípica17). También se ha observado que los miocitos de la capa no compacta no son diferentes en absoluto de los de la capa compacta, por lo que no parece una cuestión de células distintas en cada capa, sino de señalización y desarrollo de las mismas, tal como han demostrado estudios sobre el papel de la neuregulina y las vías de señalización Notch1 y ErbB2y4.
En algunas patologías, como ocurre en el síndrome de Barth18, se ha demostrado que mutaciones específicas en la tafacina pueden llevar directamente a la hipertrabeculación y no compactación del miocardio, lo que sugiere que en algunos casos la MCNC es una miocardiopatía genética definida. Sin embargo, faltan estudios que relacionen el genoma con la aparición de MCNC y desentrañen el posible papel de genes moduladores, de la epigenética o de las interacciones ambientales16.
Por otro lado, se ha visto un aumento de trabeculaciones y recesos en el VI que pueden constituir un patrón de remodelado cardíaco fisiológico por un aumento crónico de precarga, como sucede en atletas de alto rendimiento, embarazadas, afroamericanos o en la anemia drepanocítica. En estos casos, la capa compacta del corazón también es prominente19. Esta adaptación del miocardio puede regresar si desaparece la sobrecarga mecánica sobre el mismo. Por ello, también se especula con la posibilidad de que exista un sustrato genético subyacente sobre el que actuarían modificadores ambientales a la hora de desarrollar la patología, de tal forma que el cuadro podría surgir como algo adquirido con el tiempo20.
Presentación clínica
La presentación clínica es altamente variable, desde formas asintomáticas a otras muy limitantes e incluso letales11. Las tres principales complicaciones son: a) insuficiencia cardíaca (50% de los casos, con presencia de disfunción sistólica hasta en el 84% de los pacientes); b) arritmias (fibrilación auricular 25%; arritmias ventriculares 47%), y c) complicaciones tromboembólicas (0%-38% de los casos, que incluyen ictus, accidente isquémico transitorio, embolia pulmonar, isquemia mesentérica, etcétera)21.
Síntomas habituales son: disnea, edemas, palpitaciones, dolor torácico y síncope. Se ha descrito muerte súbita hasta en 18% de los casos. En la población infantil es típico un curso oscilante con una fase de recuperación después del diagnóstico seguida de un deterioro posterior.
En el electrocardiograma (ECG), hasta en 90% de los pacientes puede detectarse alguna alteración inespecífica, siendo los hallazgos más frecuentes los retrasos de la conducción ventricular, signos de crecimiento de cavidades izquierdas, anomalías de la repolarización (inversión de la onda T, cambios en el ST), ondas Q, QRS fragmentado, desviación del eje eléctrico, QT prolongado y síndrome de Wolff-Parkinson-White (0%-3% de los adultos y 9%-17% de los niños)22.
Diagnóstico
Ecocardiografía
El ecocardiograma transtorácico (ETT) es la técnica diagnóstica de primera elección para la MCNC2. Se han propuesto fundamentalmente tres criterios diagnósticos diferentes, a pesar de que no hay una definición universalmente aceptada:
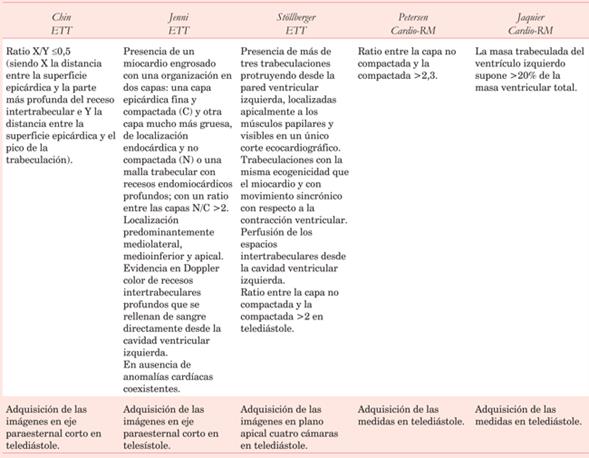
a) Criterios de Chin (1990)23: estructuración del miocardio en dos capas (una epicárdica compactada;C; y otra endocárdica no compactada;NC;) con un ratio X/Y <0,5, siendo X la distancia entre la superficie epicárdica y la parte más profunda del receso intertrabecular e Y la distancia entre la superficie epicárdica y el pico de la trabeculación. Las imágenes deben adquirirse en paraesternal eje corto y en telediástole. Tiene el inconveniente de que al tomar las imágenes en diástole, el grosor de la capa compacta es menor, lo que le confiere una mayor sensibilidad pero menor especificidad que los criterios de Jenni.
b) Criterios de Jenni o criterios suizos (2001)24: 1. Presencia de miocardio engrosado con una organización en dos capas consistente en una capa epicárdica fina y C, y otra capa mucho más gruesa de localización endocárdica y NC o una malla trabecular con recesos endomiocárdicos profundos, siendo el ratio entre las capas NC/C >2. Localización predominantemente mediolateral, medioinferior y apical; 3. Evidencia en Doppler color de recesos intertrabeculares profundos que se rellenan de sangre directamente desde la cavidad ventricular izquierda; 4. Ausencia de anomalías cardíacas coexistentes. Los criterios se aplican sobre imágenes en eje corto y en telesístole. Al realizar las mediciones en sístole, el grosor de la capa compacta se hace más prominente, lo que origina mayor especificidad y menor sensibilidad que los otros dos criterios.
c) Criterios de Stöllberger o criterios de Viena (2007)20: 1. Presencia de más de tres trabeculaciones protruyendo desde la pared ventricular izquierda; 2. Localizadas apicalmente a los músculos papilares y visibles en un único corte ecocardiográfico; 3. Trabeculaciones con la misma ecogenicidad que el miocardio y con movimiento sincrónico con respecto a la contracción ventricular; 4. Perfusión de los espacios intertrabeculares desde la cavidad ventricular izquierda; 5. Ratio entre la capa no compactada y la compactada >2 en telediástole y con adquisición de las imágenes en plano apical de cuatro cámaras.
En un estudio comparativo entre los diferentes criterios en pacientes diagnosticados de MCNC, cumplían con los criterios de Chin el 79% de los casos, con los de Jenni en el 64% y con los de Stöllberger en 53%. Los pacientes en los que coincidían todos los criterios suponían solo el 30% de la serie. Los criterios de Chin serían los más sensibles de todos ellos, pero a cambio dan mayor cantidad de falsos positivos25. Todo ello es fruto de que estos criterios han sido desarrollados en estudios con poblaciones de pocos pacientes y con un grado de reproducibilidad pobre. De hecho, con los criterios actuales, hasta 23,6% de individuos sometidos a ETT por disfunción sistólica cumplía una de las definiciones ecocardiográficas de MCNC (exceso de sensibilidad y por lo tanto tendencia al sobrediagnóstico). Incluso el 8% de controles totalmente sanos también cumplía un criterio al menos25. Además, cierto nivel de trabeculación es un constituyente normal de la anatomía del VI, así como en casos de miocardiopatía hipertrófica o hipertrofia asociada a enfermedad valvular, hipertensiva o miocardiopatía dilatada. Esto es especialmente cierto en afroamericanos y en atletas de alto rendimiento. La incertidumbre aumenta a causa de las diferencias en la definición de la trabeculación anormal y en la adquisición de planos ecocardiográficos, la mala ventana acústica, la fase del ciclo cardíaco en la que se realizan las mediciones, la subjetividad de alguno de los criterios, etcétera.
Tampoco existen recomendaciones específicas en cuanto al VD, dadas su complejidad geométrica, su estructura más trabeculada que la del izquierdo en condiciones normales y las limitaciones del ETT para definirlo adecuadamente. El empleo de técnicas ecocardiográficas avanzadas puede ayudar a definir mejor la patología en casos dudosos o de mala ventana, como ocurre con la ecocardiografía 3D, el uso de contraste o la deformación miocárdica por speckle tracking26. Esta última puede evidenciar una rotación anómala de los segmentos basales y apicales en la misma dirección, cuando lo fisiológico es que la base tenga una contracción rotacional en sentido de las agujas del reloj y el ápex antihorario. Esta técnica también ha probado ser útil para demostrar un patrón de deformación típico, con un strain rate longitudinal sistólico mayor en los segmentos basales que en el ápex, lo que puede ayudar a distinguirlo de la miocardiopatía dilatada27.
Cardio-resonancia magnética
La cardio-RM proporciona una mejor resolución espacial que el ETT, lo que ayuda a distinguir la trabeculación miocárdica y a cuantificarla, así como a obtener una mejor visualización del ápex, la pared lateral y la función cardíaca. Adicionalmente, es la técnica de elección para detectar fibrosis miocárdica y existencia de trombos intertrabeculares.
La cardio-RM debería considerarse la técnica de elección cuando la MCNC se sospecha por ETT o en casos de mala ventana acústica, o de ETT dudoso en familiares en primer grado del probando2. No está exenta, no obstante, de limitaciones como son el mayor coste y duración del procedimiento, su menor disponibilidad y los artefactos vinculados a la respiración y al movimiento cardíaco.
Los principales criterios de MCNC por cardio-RM son:
a) Criterios de Petersen (2005)28: ratio entre capa NC y C > 2,3, tomando las medidas en telediástole. Sin embargo, hay estudios que muestran que hasta 43% de individuos sanos cumplen ese límite en el eje largo (alta sensibilidad pero poca especificidad).
b) Criterios de Jacquier (2010)29: cuando la masa trabeculada del VI es >20% de la masa ventricular total, tomando las medidas en telediástole. Estos criterios más novedosos predicen MCNC con una sensibilidad y especificidad >93%, con una alta tasa de reproducibilidad interobservador, además de ayudar en la distinción del miocardio altamente trabeculado propio de la MCNC de individuos con una extensión de trabeculación normal. Sin embargo, tampoco está exenta de limitaciones, ya que estos criterios no traducen severidad de la enfermedad, solo han sido validados en sujetos caucásicos y en los cálculos se asumen como masa trabecular espacios ocupados por sangre que no constituyen masa ventricular real.
Otras técnicas diagnósticas
a) Tomografía computada cardíaca. Tiene una buena resolución espacial y capacidad para discriminar enfermedad coronaria asociada, aunque no está estandarizado su uso ni se han desarrollado criterios validados, además de los riesgos que implica el empleo de radiación ionizante y de contraste iodado, con posible deterioro de la función renal.
b) Ventriculografía izquierda. Ayuda al diagnóstico en casos de enfermedad arterial coronaria y fracción de eyección reducida. Sin embargo, tampoco existen criterios estandarizados e implica el uso de radiación y de contraste.
c) Biopsia endomiocárdica. No se recomienda su uso rutinario, pero puede ayudar en el diagnóstico diferencial de miocarditis.
Diagnóstico diferencial
Debe realizarse fundamentalmente con los siguientes cuadros: trabeculación prominente en un miocardio compacto, miocardiopatía hipertrófica (sobre todo apical), miocardiopatía hipertensiva, miocardiopatía dilatada, miocardiopatías infiltrativas, miocarditis y pericarditis, síndrome hipereosinofílico, fibroelastosis endocárdica, anomalías de las cuerdas tendinosas, trombo apical, hematoma o absceso intramiocárdico y tumores cardíacos2.
Cribado
Todo paciente con diagnóstico de MCNC debería tener seguimiento con anamnesis, exploración física, ECG, ETT y analítica con determinación de creatinkinasa en la visita inicial. En familiares en primer grado se recomienda screening clínico cada tres años desde la infancia, y si además se ha identificado una mutación genética, se recomienda screening clínico anualmente durante la infancia y cada 1-3 años en adultos2.
Estudio genético
En el probando se recomienda realizar secuenciación de ácido desoxirribonucleico (ADN) de forma dirigida en el caso de sospechar síndromes típicamente asociados a MCNC (por ejemplo, en el síndrome de Barth), o testar los principales genes asociados si no se tiene una sospecha específica. En familiares en primer grado es recomendable buscar la mutación identificada en el caso índice entre los familiares en primer grado si existiese. Sin embargo, otros autores no recomiendan rastreo de mutaciones en pacientes con MCNC a no ser que se observe un fenotipo evidente, dado que no tiene implicaciones pronósticas ni terapéuticas y no se ha establecido una clara relación causal. A pesar de todo ello, se cree que el test genético puede ayudar al diagnóstico en el 20%-40% de los pacientes30.
Tratamiento
Debe recordarse que no existen guías clínicas específicas para el manejo de la MCNC, y que éste se basa fundamentalmente en el manejo de la disfunción cardíaca o los síntomas asociados según las guías de práctica clínica generales (insuficiencia cardíaca y arritmias) y en las recomendaciones de expertos. En pacientes asintomáticos con fracción de eyección conservada se recomienda seguimiento clínico cada 2-3 años. No parece haber razones para prohibir en estos casos la práctica deportiva o el embarazo2.
En cuanto al manejo de la insuficiencia cardíaca se recomienda seguir las guías de práctica clínica habitual31, con el uso de betabloqueantes, inhibidores de la enzima de conversión (IECA) / antagonistas de receptores de angiotensina II (ARAII), diuréticos, espironolactona, sacubitrilo-valsartán, implante de desfibrilador automático (DAI)-terapia de resincronización cardíaca (TRC), inotrópicos y trasplante cardíaco llegado el caso. Hay un mayor riesgo de insuficiencia cardíaca en caso de edad avanzada, según el número de segmentos afectados, ratio NC/C elevado y presencia de realce tardío en cardio-RM.
Respecto a la prevención de complicaciones tromboembólicas, la anticoagulación en pacientes con MCNC permanece controvertida2. En los recesos intertrabeculares profundos pueden producirse remansos de sangre enlentecida que favorecen la formación de trombos. Los primeros registros tenían altas tasas de eventos embólicos (21%-38%), lo que llevó inicialmente a un manejo agresivo con empleo de anticoagulantes independientemente de la función sistólica, práctica todavía defendida por algunos clínicos32. En series más recientes, el riesgo embólico es mucho más bajo (0%-15%) en pacientes con ritmo sinusal y función normal, por lo que la anticoagulación crónica parece de más dudosa indicación. Parece recomendable la anticoagulación oral, con un objetivo de INR entre 2 y 3, en: 1. Pacientes con fibrilación auricular y/o fracción de eyección ventricular izquierda <40%; 2. Complicaciones embólicas previas; 3. Evidencia de trombo ventricular2.
Para el manejo de las arritmias cardíacas tampoco existen recomendaciones específicas por el momento y se recomienda aplicar las guías de práctica clínica general33. Aparte de esto, como recomendaciones de expertos se aboga por realizar estudio electrofisiológico en todo paciente con MCNC y arritmias sintomáticas o síncope para descartar que se puedan inducir arritmias ventriculares o supraventriculares y estudio con Holter una vez al año2. No está claro que el riesgo de arritmias en la MCNC sea superior al observado en la miocardiopatía dilatada o miocardiopatía hipertrófica. Tal como ocurre en estas, la implantación de un DAI estaría indicada en pacientes con MCNC que presenten síncope de origen no explicado, arritmias ventriculares sintomáticas, disfunción sistólica severa del ventrículo izquierdo (fracción de eyección del ventrículo izquierdo <35%) o antecedentes familiares de muerte súbita2. Tabla 1
Pronóstico
Es altamente variable, dado que los estudios iniciales arrojaban cifras de mortalidad de hasta 35% de los pacientes, mientras que estudios más recientes reportan 2%-15%2.
Los principales factores pronósticos son: 1. Edad; 2. Clase funcional; 3. Tamaño auricular; 4. Extensión de realce tardío por cardio-RM; 5. Presencia de dilatación y disfunción ventricular; 6. Arritmias ventriculares sostenidas. Al contrario de lo que se pensaba inicialmente, el ratio NC/C y el número de segmentos afectados no parecen haber demostrado implicación pronóstica2.

