











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
El síndrome de Ellis-van Creveld (EVC) es una rara anormalidad genética autosómica recesiva, caracterizada por una tétrada clásica: condrodistrofia, polidactilia postaxial, displasia ectodérmica y cardiopatía congénita. La causa del síndrome son mutaciones en los genes EVC - 1 y EVC - 2, localizados en el cromosoma 4p161,3.
Las cardiopatías congénitas ocurren en 50%- 60% de los casos y constituyen el principal determinante de la esperanza de vida. Las principales malformaciones cardíacas son: defecto del canal auriculoventricular (AV) o aurícula común, o ambos, vena cava superior izquierda persistente y anormalidades de las venas pulmonares1,4.
El síndrome fue descrito por primera vez por R. Ellis y S. van Creveld en 1940, quienes reportaron cerca de 100 casos hasta 1968; desde entonces se han reportado aproximadamente 50 casos en la literatura5, entre los cuales son escasos los pacientes adultos(2). En Sudamérica existen pocos casos descritos.
Caso clínico
Paciente masculino de 20 años, nacido en la región costa de Ecuador. Padres con cuarto grado de consanguinidad. Antecedentes prenatales: ecografía a las 36 semanas de gestación que reporta dimorfismo corporal. Antecedentes quirúrgicos: resección de un dedo supernumerario postaxial en ambas manos y colocación de material de osteosíntesis en ambas rodillas a los 9 años. Antecedentes familiares: tres primos segundos de lado paterno con discapacidad intelectual y dos primos de lado materno con acondroplasia, quienes fallecieron por causa desconocida.
Paciente asintomático que acude a control médico, donde identifican un soplo a la auscultación cardíaca, por lo que es referido a especialista para estudio. Al examen físico se encuentra: talla 133 cm, orejas de implantación baja, anodoncia parcial, paladar íntegro; elevación de hemitórax anterior izquierdo, escoliosis de columna dorsal; cicatriz de resección de dedo supernumerario y uñas hipoplásicas en ambas manos; miembros inferiores más cortos que miembros superiores, miembro inferior izquierdo acortado; rodillas en valgo (Figura 1). En la inspección de tórax se observa choque de punta en región subesternal. En la auscultación cardíaca se aprecian ruidos rítmicos, en foco mitral soplo holosistólico, regurgitante, grado III/VI, irradiado a axila; en foco pulmonar soplo sistólico eyectivo, grado II/VI, con P2 aumentado, desdoblado y fijo.
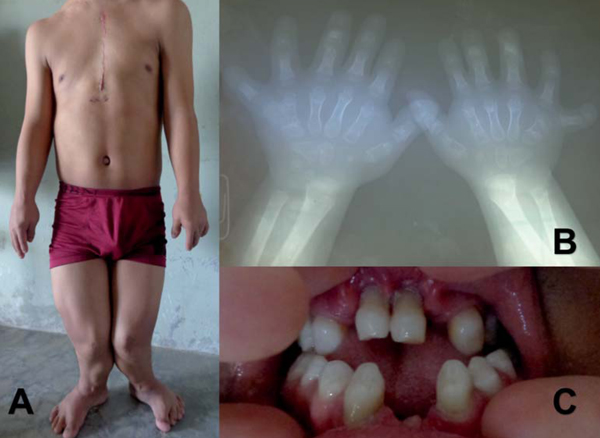
Figura 1: A. Paciente luego de corrección quirúrgica de defecto cardíaco (esternotomía media), características clínicas descritas en el texto. B. Dedos supernumerarios en la niñez, antes de su resección. C. Anodoncia parcial.
En el electrocardiograma destaca PR prolongado, bloqueo completo de rama derecha y hemibloqueo anterior izquierdo. En la radiografía de tórax se evidencia hiperflujo pulmonar y escoliosis de la columna dorsal con concavidad izquierda (Figura 2). El ecocardiograma transtorácico reportó comunicación interauricular tipo ostium primum de 22 mm, comunicación interventricular de 15 mm totalmente ocluida por velo septal tricuspídeo convirtiendo un canal AV intermedio en un canal AV parcial e hipertensión pulmonar leve.
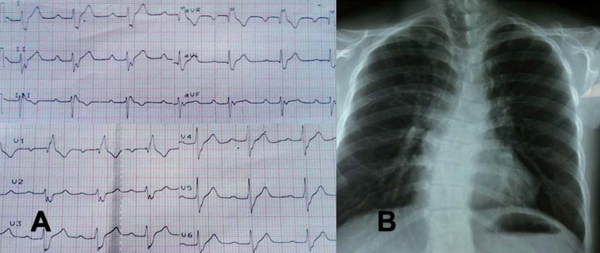
Figura 2: A. Electrocardiograma, aVR, aVL y aVF registradas a 1mV/5mm: Ritmo sinusal, 75 lpm, Eje QRS - 90°, intervalo PR de 240 ms, complejo QRS de 120 ms con morfología de bloqueo completo de rama derecha y hemibloqueo anterior izquierdo. B. Radiografía A-P: Situs solitus, índice cardiotorácico de 0,48, cono de arteria pulmonar prominente, aumento de la trama vascular, escoliosis de la columna dorsal.
Por las características clínicas es diagnosticado como EVC. El paciente es hospitalizado para corrección quirúrgica del defecto cardíaco. Hallazgos quirúrgicos: comunicación interauricular tipo ostium primum de 5 cm2; ausencia de cleft mitral; ausencia de comunicación interventricular. Es realizada atrioseptoplastia con parche pericárdico bovino. El paciente recibe el alta hospitalaria después de seis días, sin complicaciones.
Discusión
La prevalencia exacta de EVC es desconocida5; se estima una incidencia en recién nacidos de 1/60.000, siendo más común en algunas poblaciones como la comunidad Amish de Lancaster County, Pennsylvania, en Estados Unidos, con una incidencia estimada de 5/1.0006. No se encontraron casos clínicos reportados en Ecuador. Se desconoce la existencia de casos similares en la comunidad de nacimiento del paciente.
El diagnóstico prenatal se realiza con ecografía desde el primer trimestre, observando engrosamiento de la translucencia nucal, polidactilia postaxial, tórax estrecho, huesos largos arqueados y acortados, defectos cardíacos, entre otras alteraciones estructurales5-7. A pesar de que el paciente fue evaluado mediante ecografía prenatal y posteriormente intervenido quirúrgicamente en la niñez, el diagnóstico de EVC no fue efectuado. Es importante recalcar que la baja sospecha clínica de cardiopatías congénitas afecta directamente su manejo y pronóstico.
Las características clínicas típicas del síndrome son condrodistrofia, con una talla promedio en el adulto entre 109 y 155 cm; polidactilia, afectando principalmente las manos, comúnmente ambas, y en raras ocasiones los pies; displasia ectodérmica, presentándose como anormalidades en dientes (erupción tardía, anodoncia parcial) y uñas (hipoplásicas, displásicas, friables, ausentes), y anormalidades cardíacas, siendo principalmente afectada la aurícula. El desarrollo cognitivo es normal5,6,8. En este paciente encontramos la tétrada clásica, por lo que es posible realizar el diagnóstico clínico de EVC (Tabla 1).
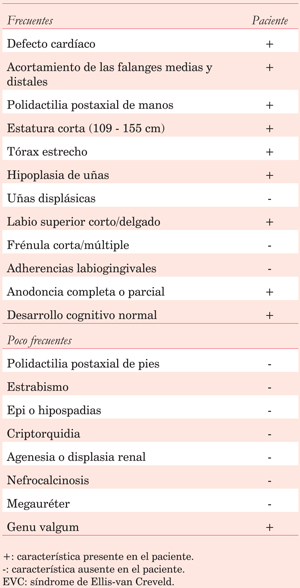
Uno de los principales diagnósticos diferenciales lo constituye la disostosis acrofacial tipo Weyer, una condición autosómica dominante, donde coexisten anomalías dentales, polidactilia postaxial de manos y pies, estatura corta; pero no existe cardiopatía congénita ni tórax estrecho2,6. Otro diagnóstico diferencial es el síndrome de Jeune, condición autosómica recesiva, caracterizada por tórax estrecho, acortamiento de extremidades, displasia ósea, anomalías renales y degeneración retiniana, pero algunas características como la cardiopatía congénita o la displasia ectodérmica apoyan al diagnóstico de EVC2,5.
El manejo de EVC es multidisciplinario; el adecuado control de los defectos cardíacos define su pronóstico1,5. En este paciente los defectos cardíacos fueron resueltos a edad adulta debido al diagnóstico tardío. La resolución quirúrgica fue satisfactoria.
Es crucial insistir en la importancia del diagnóstico temprano, no solo de EVC, sino de todas las cardiopatías congénitas, idealmente en la etapa prenatal. Es necesario brindar mayor información a la comunidad médica sobre este tipo de síndromes para facilitar su diagnóstico clínico.

