











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
Las miocardiopatías son un grupo de enfermedades del miocardio, con características comunes y un abordaje clínico particular. Entre las principales miocardiopatías encontramos1:
- Miocardiopatía hipertrófica (MCH).
- Miocardiopatía dilatada (MCD).
- Miocardiopatía arritmogénica (MCA).
- Miocardiopatía restrictiva (MCR).
- Miocardiopatía no compactada (MCNC).
Este grupo de patologías explica una proporción importante de las muertes súbitas en individuos menores de 35 años de edad2,3. Son clínicamente muy heterogéneas, lo que dificulta el diagnóstico, principalmente en etapas incipientes, y representan un desafío para la estratificación de riesgo4. Tienen una base genética que se traduce en la importancia de realizar una evaluación familiar en busca de otros afectados y posiciona el estudio genético como una prueba complementaria relevante5.
Gracias a los últimos avances en los métodos de secuenciación del ácido desoxirribonucleico (ADN), los estudios genéticos se han hecho más accesibles y han demostrado gran utilidad en el diagnóstico, manejo clínico y estratificación pronóstica de las miocardiopatías. Tanto es así, que la mayoría de las guías internacionales y consensos de expertos incluyen esta herramienta entre sus recomendaciones. Abordaremos la utilidad clínica práctica del estudio genético en las miocardiopatías.
Conceptos básicos para la aplicación de la genética en las miocardiopatías
Las miocardiopatías son generalmente enfermedades monogénicas, donde la presencia de una mutación puede ser causa necesaria y suficiente para el desarrollo del fenotipo. Es importante aclarar algunos conceptos básicos relacionados con la genética de las miocardiopatías que facilitarán la interpretación y aplicación de los resultados de este estudio complementario en la práctica clínica.
Patrón de herencia: es la forma en la que se transmite la mutación o variante genética entre los integrantes de una familia. La mayor parte de las miocardiopatías siguen un patrón autosómico dominante, esto significa que la presencia de la mutación en uno de los alelos del gen es suficiente para desencadenar la enfermedad. Además, las mutaciones afectan a genes de los cromosomas somáticos (autosomas), que pueden transmitirse desde/hacia hombres y mujeres indistintamente, con una probabilidad del 50% en cada gestación. En algunos casos, los genes involucrados están en el cromosoma X y las mutaciones siguen un patrón de herencia particular (ligado a X), donde tanto mujeres como hombres pueden transmitirlo (estos últimos solo a sus hijas), y los hombres portadores suelen desarrollar la enfermedad en forma más precoz o severa que las mujeres. En una minoría el patrón de herencia es autosómico recesivo, siendo necesaria la presencia de ambos alelos del gen mutados para el desarrollo del fenotipo (homocigotos o heterocigotos compuestos); los progenitores que solo tienen un alelo mutado están libres de enfermedad (la consanguinidad en la familia facilita este tipo de patologías).
Heterogeneidad genética: significa que cada enfermedad está causada por un número variable de mutaciones en genes diferentes. En algunos casos esta heterogeneidad es muy grande, como ocurre en la MCD donde se han descrito más de 100 genes relacionados. El gran avance reciente en las tecnologías de secuenciación permite realizar estudios mediante paneles que incluyan todos estos genes6.
Heterogeneidad fenotípica: este concepto hace referencia a que mutaciones en un mismo gen (o incluso la misma mutación) pueden estar relacionadas con fenotipos diferentes. Por ejemplo, las mutaciones en el gen MYH7 (codifica la beta-miosina cardíaca) son una de las causas principales de la MCH, pero también se han relacionado con la MCD, la MCNC y la MCR7.
Penetrancia: representa la proporción de portadores de una mutación que han desarrollado el fenotipo a una determinada edad. En las miocardiopatías la penetrancia es incompleta en casi todos los casos y habitualmente aumenta con la edad. Este concepto es importante a la hora del asesoramiento genético familiar, ya que el ser portador de una mutación no significa que inexorablemente se vaya a desarrollar la enfermedad8.
Expresividad: mientras que la penetrancia es una variable dicotómica (se desarrolla o no el fenotipo), la expresividad es un fenómeno continuo y que hace referencia al grado de severidad de las manifestaciones clínicas. También es muy variable en las miocardiopatías y puede depender de múltiples factores modificadores, ya sea ambientales o genéticos. De esta forma, es relativamente común que, dentro de una misma familia, los portadores de una determinada mutación expresen grados variables de afectación clínica, desde individuos con alteraciones sutiles o fenotipos borderline hasta sujetos con patologías severas y precoces9.
Cosegregación: hace referencia a la comparación de los resultados del estudio clínico y genético en los integrantes de una familia. Permite asegurarse que una determinada variante genética es la causa de la enfermedad, especialmente cuando la información previa disponible no es suficiente para definirlo. La cosegregación se cumple cuando todos los familiares clínicamente afectados son portadores de la mutación y los no portadores están sanos. La excepción a esta regla son aquellos casos o familias donde hay más de una mutación causal. Por esta última razón, y para no descartar variantes genéticas relevantes durante el estudio de cosegregación, resulta muy importante la correcta selección del individuo por el que comenzar el estudio genético en la familia.
Abordaje práctico de una familia
En general, el primer individuo que se diagnostica dentro de una familia se denomina caso índice o probando. A partir de su diagnóstico, se debe realizar una evaluación clínica en cascada de los familiares, empezando por los de primer grado (progenitores, hermanos, hijos). Estas revisiones deben comenzar en etapas precoces de la vida, habitualmente en la infancia, y repetirse periódicamente hasta avanzada la edad adulta (60-65 años de edad). Esta estrategia permite detectar precozmente manifestaciones del fenotipo en familiares y aplicar a tiempo recomendaciones sobre el estilo de vida o tratamientos10).
Cuando nos disponemos a realizar un estudio genético en una miocardiopatía, deberemos considerar que los resultados son importantes para toda la familia. Por ello, es imprescindible seleccionar correctamente el individuo más adecuado para lograr el mayor rendimiento. Este paciente suele ser el caso índice, a menos que dentro del estudio clínico en cascada se identifique otro familiar con un fenotipo mucho más severo o precoz; en este caso, optaremos por comenzar el estudio por este individuo11. Estas patologías siguen generalmente un patrón monogénico, en donde la presencia de una única variante genética (o mutación) es el determinante necesario principal de la patología. Sin embargo, en un porcentaje variable de familias (habitualmente entre el 5% y 7%, pero puede ascender hasta el 40% en MCA) el estudio genético identifica más de una variante causal, lo que en algunos casos explica distintos grados de severidad en la expresión clínica9. Comenzar el estudio por aquel familiar más severamente afectado es una forma de evitar diagnósticos genéticos incompletos que lleven a errores en la aplicación de los resultados12. Será de suma importancia intentar preservar una muestra de material genético de aquellos pacientes que sufran una muerte súbita y cuya autopsia oriente hacia la presencia de una miocardiopatía13.
Una vez correctamente seleccionado el caso a estudiar genéticamente, tendremos que escoger el tipo de análisis más adecuado. Actualmente disponemos de paneles que incluyen todos los genes con evidencia etiológica científicamente demostrada para las diferentes miocardiopatías. Se seleccionará aquel panel correspondiente al diagnóstico clínico del paciente. En el caso de fenotipos complejos, solapados, o cuando hay datos clínicos de distintas patologías en la familia, se puede optar por paneles generales que incluyen genes relacionados con distintas patologías (por ejemplo, panel general de miocardiopatías).
Indicaciones del estudio genético en miocardiopatías
Las distintas guías de práctica clínica internacionales han posicionado al estudio genético como una recomendación clase I para la mayoría de estas enfermedades13-17. En consecuencia, la indicación de un estudio genético se encuentra justificada en cualquier paciente con un diagnóstico clínico confirmado. De todas formas, vamos a enumerar distintas situaciones y matices de la indicación de un estudio genético.
- Paciente con diagnóstico clínico confirmado: el estudio genético permite realizar un diagnóstico etiológico preciso que repercutirá en un manejo clínico adecuado del paciente y en la estratificación pronóstica. Identificar la mutación causal en la familia permitirá detectar precozmente familiares en riesgo; este aspecto será abordado en detalle más adelante.
- Paciente con sospecha diagnóstica o fenotipo borderline: la identificación de una mutación claramente patogénica forma parte de los criterios diagnósticos en distintas cardiopatías familiares ( MCA, por ejemplo). En algunos casos, la presencia de una mutación permite hacer un diagnóstico diferencial con adaptaciones fisiológicas del corazón (por ejemplo, la diferenciación entre el corazón de atleta y una MCH incipiente)18,19.
- Paciente con muerte súbita/arritmia ventricular de causa desconocida: el estudio genético debe formar parte del algoritmo diagnóstico en pacientes jóvenes (hasta 35 años). En mayores también debe considerarse una vez descartada la cardiopatía isquémica (principal sustrato en este grupo etario)13.
Rentabilidad del estudio genético
Se entiende por rentabilidad el porcentaje de estudios genéticos que resultan positivos; es decir, que son capaces de identificar una mutación causal. Una premisa importante a tener en cuenta es que la rentabilidad será limitada y el porcentaje dependerá del tipo de miocardiopatía. En general, la rentabilidad del estudio genético para la mayoría de las miocardiopatías es de 40%-60%4,5,11,14.
Sin embargo, no hay que olvidar que el análisis genético es un estudio complementario más en el arsenal diagnóstico. En consecuencia, su rentabilidad depende directamente de la probabilidad pretest clínica de cada paciente en particular. Es decir, cada miocardiopatía puede tener un estimado de rentabilidad del estudio genético considerando el global de pacientes con el mismo diagnóstico, pero las características clínicas y familiares de cada caso pueden hacer que la rentabilidad sea mayor o menor que lo esperable para su cardiopatía20.
Un estudio genético negativo no cuestiona el diagnóstico clínico de la enfermedad, ni descarta la presencia de una patología genética y hereditaria. Muchas son las razones que pueden explicar un resultado negativo: por un lado, el conocimiento sobre el sustrato genético en estas patologías se encuentra en evolución constante y algunos de los genes están aún por descubrirse. También es posible que los métodos de secuenciación actuales tengan limitaciones en identificar determinadas mutaciones (regiones intrónicas profundas, mutaciones especiales, etc.)21. Finalmente, existe la posibilidad de que en algunos casos los desencadenantes sean múltiples o relacionados con causas no genéticas.
Interpretación de un estudio genético
Los nuevos métodos de secuenciación masiva han resuelto muchas limitaciones técnicas del estudio para laboratorios con experiencia y que adoptan los estándares internacionales22. Por el contrario, la dificultad en la interpretación de los resultados y la categorización de las variantes genéticas es cada vez más dificultosa. Este paso es de vital importancia dentro de un estudio y depende en gran medida de la experiencia del centro que realiza la interpretación23. La misma debe ser realizada por un equipo multidisciplinario que incluya bioinformáticos, biólogos, genetistas, y, sobre todo, cardiólogos. La interpretación final de un estudio genético debe ser eminentemente clínica y personalizada para cada paciente en particular.
Categorización de la patogenicidad de una variante
La posibilidad de incorporar múltiples genes en el análisis permite identificar una gran cantidad de variantes que deben ser clasificadas e interpretadas. La gran mayoría no tendrá ninguna relevancia y la tarea más importante consistirá en identificar aquella variante potencialmente causal de la enfermedad y su grado de asociación con la patología de acuerdo a la evidencia disponible (Tabla 1). Para ello, se toman en cuenta distintos factores clínicos y funcionales.
Tabla 1: Clasificación de las variantes genéticas de acuerdo a la información disponible y utilidad clínica que se puede obtener de cada una de ellas. Modificada de Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology24.

- Tipo de variante genética y gen afectado: en líneas generales, las variantes que producen un codón de stop prematuro o aquellas que afectan al proceso de corte y empalme del ácido ribonucleico mensajero (ARNm) (variantes de tipo “truncamiento”) suelen ser consideradas potencialmente más deletéreas; dentro de este grupo de variantes “radicales” también podríamos incluir a las grandes deleciones o duplicaciones del ADN. Le siguen las variantes de tipo “missense”, en donde el cambio de un nucleótido genera el cambio de un aminoácido por otro; en el mismo nivel podríamos incluir a aquellas deleciones o inserciones de un (o unos pocos) aminoácidos. Por último, las variantes sinónimas (existe cambio de un nucleótido pero no cambia el aminoácido original) o intrónicas, en donde no hay cambio de la estructura proteica, serían inocuas siempre y cuando no afecten el corte y empalme del ARNm.
- Frecuencia poblacional de la variante: cuanto más frecuente sea una variante en la población “control” (individuos no afectados de la población general), menor será la probabilidad de que sea patogénica. Las miocardiopatías tienen una prevalencia baja en la población general (la más frecuente es la MCH, que afecta 1/500 individuos); difícilmente una variante genética puede ser patogénica cuando su frecuencia en controles excede la frecuencia de la propia enfermedad25.
- Fenotipo del paciente en particular versus fenotipo descrito para ese tipo de variantes en el gen: no es lo mismo una variante “missense” no descrita en el gen GLA en un paciente que presenta una MCH de tipo concéntrica, se encuentra en diálisis por una insuficiencia renal crónica, con una historia de dolor neuropático y angioqueratomas, que el mismo hallazgo en un paciente joven con MCD y disfunción sistólica severa. En el primer caso la probabilidad de que la variante en el gen GLA (que codifica la enzima alfa-galactosidasa) explique el fenotipo (enfermedad de Fabry) pasa a ser muy elevada; por el contrario, la posibilidad de que explique el fenotipo del segundo caso es muy baja.
- Descripciones previas de la variante en pacientes; cosegregación con un fenotipo determinado en una familia o presentación “de novo”: la probabilidad de que una variante sea patogénica es mayor cuantas más veces haya sido identificada en pacientes asociada a un fenotipo particular. De la misma manera, la cosegregación de la variante con un fenotipo en una familia con un número suficientemente grande de individuos evaluados es un criterio de mucho peso en la patogenicidad (la mayoría de los portadores de la variante se encuentran afectados, mientras que los no portadores no desarrollan el fenotipo). La presencia de una mutación “de novo” (ambos progenitores son no portadores de la variante y se encuentran sanos, mientras que el descendiente afectado es portador) es otro indicador de patogenicidad; la presentación “de novo” suele ser bastante frecuente en algunas RASopatías (mutaciones en componentes de la vía de transducción de señal RAS-MAPK (proteincinasas activadas por mitógenos) como el síndrome de Noonan, que puede tener como manifestación cardiovascular la presencia de MCH o estenosis de la arteria pulmonar, o ambas.
- Otros criterios de soporte: dentro de estos incluimos algunas herramientas bioinformáticas que tienen en cuenta características evolutivas, de conservación, y físico-químicas particulares de cada variante para predecir que ésta sea deletérea o tolerada. Si bien son muy utilizadas, no deben ser la única característica a tener en cuenta a la hora de definir la patogenicidad y su utilidad es limitada en nuestra opinión.
Resultado de un estudio genético
Cuando recibimos un estudio genético nos enfrentamos a tres grandes posibilidades (Figura 1).
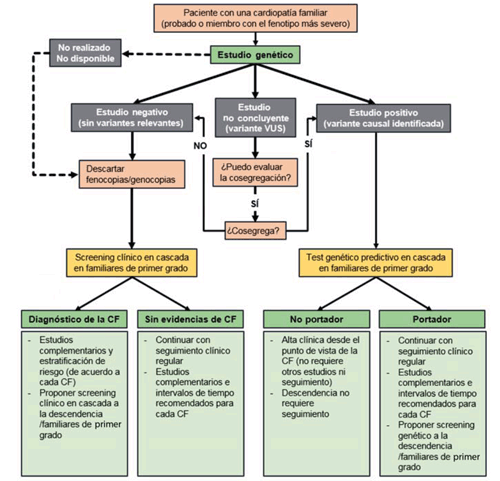
Figura 1: Algoritmo diagnóstico y del manejo de los pacientes y familiares tras la realización de un estudio genético en un probando con diagnóstico de una cardiopatía familiar. CF: cardiopatía familiar
- Estudio genético positivo: se ha identificado una variante patogénica o muy posiblemente patogénica que explica la enfermedad.
- Estudio genético no concluyente: se ha identificado una variante de significado clínico incierto (VUS es su acrónimo en inglés: variant of unknown significance), o que podría ser la causa del fenotipo pero la información actual no permite confirmarlo.
- Estudio genético negativo: no se han identificado variantes que expliquen la enfermedad en el caso estudiado.
Estudio genético positivo
En este caso se ha identificado una (o más de una) variante genética que explica el fenotipo del paciente. La identificación de una variante patogénica o muy probablemente patogénica tiene importantes implicaciones y valor agregado para el manejo del paciente y la familia.
Diagnóstico: la identificación de una mutación causal es la que confirma el diagnóstico etiológico de la enfermedad. Como ya fue mencionado previamente, la presencia de una mutación forma parte incluso de los criterios diagnósticos de algunas cardiopatías familiares. Un fenotipo particular es la vía final común, pero que a nivel molecular puede tener etiologías muy diversas. Pongamos como ejemplo la miocardiopatía familiar: la dilatación y disfunción del ventrículo izquierdo es la última manifestación de un proceso que empezó mucho antes, y que además estaba determinado genéticamente desde la concepción. Además, este fenotipo puede estar causado por mutaciones en genes que codifican proteínas localizadas en el sarcómero cardíaco (MYH7, MYBPC3, TNNT2)26, filamentos intermedios (FLNC, DES)27,28, proteínas de la membrana nuclear (LMNA)29, u otras que participan de vías que incluyen chaperonas y la modulación del corte y empalme del ADN (BAG3, RBM20)30,31. Como veremos posteriormente, tanto el cuadro clínico y el pronóstico están determinados genéticamente y no es igual a pesar de que todas puedan expresar un fenotipo relativamente similar.
Valor clínico predictivo del test genético o utilización de una variante como test predictivo en el screening familiar: este concepto proviene de la posibilidad que existe de realizar un test a un individuo que se encuentra en un estadio presintomático (todavía no ha desarrollado la enfermedad), pero que debido a ser portador de una mutación causal que ha sido identificada previamente en la familia, se encuentra en riesgo de desarrollar la enfermedad en el futuro11.
En la mayoría de las miocardiopatías la penetrancia de la enfermedad depende de la edad. Es decir, el riesgo de desarrollar un fenotipo evidente aumenta a medida que el individuo envejece. En líneas generales, y tomando a la MCH como ejemplo, es muy raro que los portadores de una determinada mutación desarrollen hipertrofia a edades tempranas, aumentando la misma con la edad (Figura 2). En estos casos, identificar un portador que aún no ha desarrollado el fenotipo a los 20 años es importante, ya que este individuo está en riesgo de desarrollar la enfermedad en el futuro (de hecho, solo un porcentaje muy pequeño no la hará durante toda su vida). O expresado de otra manera, en este caso un test genético positivo es predictivo de enfermedad en el futuro y la presencia de la variante genética puede ser utilizada con fines predictivos en individuos asintomáticos o no afectados.
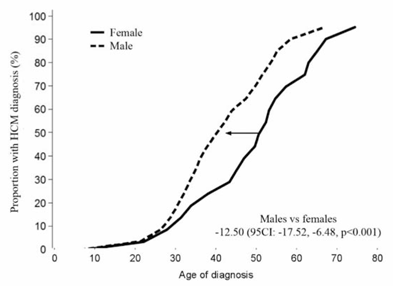
Figura 2: Proporción de individuos con diagnóstico de MCH portadores de variantes patogénicas en los genes MYBPC3 y MYH7. La incidencia de MCH comienza a aumentar luego de los 20-30 años, siendo excepcional por debajo de los 10 años, llegando a ser mayor a 90% a los 70 años. También puede observarse cómo las mujeres se diagnostican unos diez años más tarde que los varones, presentando una penetrancia menor de la enfermedad a cualquier edad (p< 0,001). La mediana de la edad de diagnóstico (edad a la que el 50% de los portadores presentaban un diagnóstico de MCH) fue de aproximadamente 40 años en hombres y 50 años en mujeres. Gentileza de I. Pérez-Sánchez, et al32.
El screening familiar: la identificación de una variante que es causa de enfermedad y puede utilizarse como test predictivo en el screening familiar es probablemente la mayor utilidad de la realización de un test genético en una familia. El patrón de herencia en la mayoría de las miocardiopatías es autosómico dominante, por lo que cada familiar de primer grado del probando tiene un 50% de posibilidades de ser portador de la variante. A menos que la variante tenga una presentación “de novo”, habrá sido heredada de uno de los dos progenitores, y cada uno de los hermanos tiene un 50% de posibilidades de ser portador, al igual que lo es el riesgo de transmitirlo a la descendencia. El estudio genético se convierte en la herramienta más potente a la hora del screening familiar, ya que permite identificar aquellos familiares en riesgo que requerirán seguimiento clínico y dar de alta a los no portadores de la variante causal33,34).
Estudio genético no concluyente
En algunas ocasiones, el estudio genético identifica variantes de significado incierto o desconocido (VUS)35. Esto ocurre cuando la evidencia disponible no es suficiente para establecer una asociación causal, ni para descartarla y considerarla una variante benigna. Este resultado no permite una actuación clínica directa sobre el paciente y su familia. En general, estas variantes están ausentes o tienen una frecuencia extremadamente baja (< 0,1%) en la población general. Las VUS suelen presentar una o más de estas características:
- No han sido previamente identificadas en otros pacientes afectados.
- Están descritas en un número muy limitado de casos aislados, pero no hay datos que demuestren su cosegregación familiar.
- Afectan a genes relacionados con la patología pero con un nivel de evidencia bajo por el momento.
- Están presentes en la población general con una frecuencia que excedería la prevalencia general de la enfermedad.
- Su mecanismo funcional es diferente al esperado para variantes patogénicas en el mismo gen, o no está claramente demostrado que el cambio a nivel genético se traduzca en una alteración proteica.
Esta categoría de variantes puede ser muy amplia y en muchos casos será necesario matizar nuestra interpretación hacia un resultado que puede ser clínicamente relevante, o no tanto. Es sumamente importante la experiencia clínica del equipo o centro que realiza el informe genético para intentar reducir al máximo la incertidumbre en este tipo de resultados.
Utilidad y forma correcta de realizar el estudio de cosegregación familiar ante un estudio genético de significado incierto
Evaluar la cosegregación familiar puede ser una tarea muy importante para definir la patogenicidad de una variante genética de significado incierto. Para realizarlo correctamente, el primer paso es evaluar clínicamente la mayor cantidad de familiares posibles. Posteriormente, se analizará la presencia de la variante en estudio tanto en familiares sanos como en aquellos clínicamente afectados. Es muy importante incluir a los progenitores o las generaciones más añosas dentro de una familia, ya que pueden aportar información muy valiosa en el estudio de cosegregación. El hecho de identificar la variante en un familiar que se encuentra clínicamente sano con más de 75-80 años de edad disminuye las posibilidades de que sea la única causa genética de la enfermedad en la familia (aunque no lo descarta completamente, basados en el concepto de penetrancia incompleta), especialmente si el probando se presentó con un fenotipo severo y precoz. Una situación que es determinante y descarta la asociación directa de una variante con el fenotipo, es su ausencia en un familiar claramente afectado por la enfermedad. Cuantos más individuos de distintas generaciones, tanto sanos como enfermos, incluyamos en el estudio de cosegregación, más robustos serán los fundamentos para reclasificar la variante. Hasta llegar a conclusiones válidas, los resultados deben analizarse en contexto de investigación y no permitirán tomar decisiones clínicas. Sin embargo, en muchos casos se demuestra una asociación contundente de la variante con el fenotipo dentro de la familia, que es suficiente para aumentar la patogenicidad y utilizarla con fines predictivos (como se comentó previamente).
Estudio genético negativo
Son aquellos análisis que no son capaces de identificar una causa genética potencial para la enfermedad del paciente. En contra de lo que habitualmente se cree, esta situación es la menos deseable para el paciente y su familia. Por un lado, no es posible resolver la incertidumbre sobre la causa específica de la enfermedad en el caso índice ni sumar información a la estratificación pronóstica. Por otro lado, un resultado negativo no descarta la presencia de la patología ni su carácter genético y hereditario. Son estudios que no permiten seleccionar los integrantes de una familia que realmente se beneficiarán de las revisiones clínicas periódicas ni interrumpir el seguimiento en aquellos que no están genéticamente predispuestos. En consecuencia, todos los familiares de primer grado deberán someterse a evaluaciones clínicas con intervalos de tiempo variables, hasta edades avanzadas. En general, para las miocardiopatías se considera que estas revisiones deben comenzar en la infancia (alrededor de los 8-10 años de vida) y repetirse periódicamente (más frecuente durante la adolescencia y la etapa adulto-joven) hasta la sexta o séptima década de vida34.
Aspectos a evaluar ante un resultado negativo del estudio genético
¿El estudio genético fue adecuado e interpretado por personal idóneo?
Deberemos analizar si todos los genes relacionados con el fenotipo del paciente han sido incluidos en el estudio. El avance en el descubrimiento de nuevos genes en miocardiopatías es continuo. Es por ello importante considerar el nivel de actualización del panel utilizado en el estudio. Determinados laboratorios no realizan una revisión periódica y sistemática de la literatura para incorporar nuevos genes a sus paneles. Por otro lado, el grado de conocimiento y experiencia clínica del equipo que realiza la interpretación e informe es fundamental a la hora de evitar descartar variantes que pueden ser relevantes.
¿Se secuenciaron en forma completa y con buenas coberturas los genes del panel?
Puede haber mucha variabilidad en la calidad de secuenciación que ofrecen centros diferentes. Es importante evaluar si los genes del panel fueron analizados completos (todas las regiones codificantes y regiones intrónicas flanqueantes), ya que las estrategias de algunos centros es evaluar en determinados genes solo las regiones donde se han descrito previamente mutaciones. La cobertura o profundidad de lectura hace referencia a la cantidad de veces que se ha leído una determinada posición genómica, y es fundamental para evitar falsos negativos en la detección de variantes.
¿Se analizaron CNVs (copy number variations) en aquellos genes donde son frecuentes este tipo de mutaciones?
En general, más del 95% de las variantes genéticas asociadas a miocardiopatías son cambios puntuales de un nucleótido por otro, o pequeñas inserciones/deleciones de un número limitado de nucleótidos dentro de la secuencia (menos de 20 en su mayoría). Sin embargo, algunos genes tienen mayor tendencia a presentar mutaciones que suponen la deleción o duplicación de grandes regiones genómicas (uno o más exones completos, que pueden abarcar kilo o megabases). Este tipo de variantes (denominadas genéricamente CNVs) se detectan con un abordaje técnico específico y distinto al utilizado para identificar cambios puntuales o pequeñas indels, que muchas veces se ofrece como una técnica adicional. Sin embargo, en algunos centros se ha perfeccionado la técnica de secuenciación masiva en paralelo (NGS) para detectar este tipo de variantes en el mismo estudio. A modo de ejemplo, la MCD secundaria a mutaciones en distrofina (gen DMD) es debida a este tipo de CNVs en un 70%-80% de los casos. Un estudio genético negativo en un paciente con una sospecha diagnóstica de esta patología que no haya evaluado la presencia de CNVs en DMD supondrá una limitación grande, con muchas probabilidades de ser un falso negativo36).
¿Realmente mi paciente tiene el fenotipo? Probabilidad pre y postest. Considerar otros diagnósticos/genocopias
Finamente, una vez consideradas todas las cuestiones anteriores, deberemos preguntarnos si el paciente tiene un fenotipo claramente compatible con una miocardiopatía. Los diagnósticos clínicos limítrofes o borderline, en presencia de otros factores de confusión (como la hipertensión arterial en la MCH) tienen una probabilidad pretest más baja de que el estudio genético sea positivo20. En estos casos, posiblemente no sea necesario o costo-efectivo plantearse estudios genéticos más complejos o de ampliación. Una situación muy distinta es el estudio genético negativo cuando la probabilidad pretest es alta y la patología tiene una presentación claramente familiar con varios miembros afectados. En estos casos, se podría plantear la realización de un exoma (estudio de las regiones codificantes de todos los genes; alrededor de 25.000) o un genoma (estudio de todo el ADN del código genético humano), en busca de variantes en genes no asociados con la patología hasta el momento. Estas estrategias tienen más rendimiento cuando se las aborda en formas de tríos (estudio del probando y sus progenitores al mismo tiempo) o cuartetos, incluyendo familiares afectados y sanos. Para el filtrado de variantes candidatas se tendrá en cuenta el patrón clínico de herencia que parece seguir la patología en la familia, comparando los resultados de genotipo y fenotipo en cada familiar. Sin embargo, estas estrategias caen en el terreno exclusivo de la investigación. El hecho de que una variante en un gen no asociado previamente con una determinada patología se considere causal, requerirá de múltiples estudios (funcionales y clínicos) antes de poder utilizar este resultado en un contexto clínico.
Valor agregado de un estudio genético en las miocardiopatías para el manejo del paciente y la familia
1) Costo-efectividad
La gran ventaja de incluir el estudio genético dentro de los estudios complementarios es que cuando detectamos la variante genética que es la causa de la enfermedad en el probando de la familia, podemos utilizar la misma como estrategia de screening. Es decir, evaluar a todos los familiares de primer grado no solo clínica, sino genéticamente para ver si son portadores o no portadores de la variante en cuestión11. Es importante remarcar en este punto que en el caso índice el costo del estudio será superior, ya que debemos realizar un estudio amplio mediante NGS que incluya un número elevado de genes, mientras que en los familiares solo iremos a buscar la variante que es la causa de la enfermedad de la familia mediante una técnica más barata (en general, secuenciación de tipo Sanger). Podemos considerar el estudio del caso índice (más caro) como una inversión para el futuro, ya que tendrá implicaciones en toda su familia y descendencia23.
La identificación de una variante en el probando que puede utilizarse con valor predictivo nos permite dividir a los familiares en dos grupos.
- Familiares portadores: son los individuos que se encuentran en riesgo, en general en una fase preclínica de la enfermedad. Aquí es donde hay que poner los recursos del sistema para el seguimiento de los mismos. Cuanto más sepamos de la variante genética en cuestión, más podremos predecir acerca del probable curso clínico de la enfermedad y del pronóstico (edad de presentación del fenotipo, características clínicas, eventos, comportamiento en hombres versus mujeres, etc.) y anticiparnos a las posibles complicaciones.
- Familiares no portadores: en estos individuos la causa genética de la enfermedad en la familia se encuentra ausente, pudiéndose interrumpir los seguimientos clínicos.
Este modelo es el utilizado actualmente y ha demostrado ser costo-efectivo37. Es decir, es más barato para el sistema de salud afrontar los costos de realizar estudios genéticos (inclusive cuando no todos ellos serán positivos) para poder discriminar qué familiares estarán en riesgo y cuáles no, que solamente utilizar la estrategia de seguimiento clínico en todos los familiares de primer grado. Se han realizado estudios en donde, incluso con rentabilidades diagnósticas muy bajas (identificando variantes predictivas en menos del 40% de los casos), la costo-efectividad se inclinaba hacia la estrategia que utilizaba el diagnóstico genético38.
2) Valor pronóstico
Existen discrepancias en la literatura acerca del valor pronóstico que puede aportar la genética en las miocardiopatías39. En este sentido, nuestro grupo ha sido pionero y tenemos una opinión formada, la que coincide con la de otros líderes europeos: no existen dudas de que uno de los valores agregados de la genética es la posibilidad de brindar información pronóstica. Es real que esto no es posible en todos los casos estudiados, pero a medida que vamos avanzando en la comprensión de las enfermedades cardíacas hereditarias y más información se reúne acerca de las mutaciones causales, mayores son los casos en que es posible establecer un pronóstico con un margen razonable.
En esta sección utilizaremos como ejemplo la MCH, tal vez el paradigma de las miocardiopatías hereditarias, la más frecuente, y en la que existen ciertas discrepancias acerca de la utilidad pronóstica (fundamentalmente basada en la opinión de algunos grupos americanos)40,41. Para esto, utilizaremos los datos provenientes de una base de datos en donde se ha recogido la información clínica de los portadores de mutaciones reportados en la literatura, sumados a los que hemos genotipado en nuestro centro de los que dispongamos de edad de último seguimiento o del evento cardiovascular. Las curvas se han construido desde el nacimiento mediante el método de Kaplan-Meier, y se evalúa la sobrevida libre de muerte cardiovascular (definida como muerte súbita, descarga apropiada de cardiodesfibrilador implantable, muerte por fallo cardíaco, trasplante y muerte por otra causa cardiovascular o periprocedimiento).
En la Figura 3 puede observarse la curva de sobrevida en portadores de variantes genéticas patogénicas en tres genes cuyo mecanismo molecular es totalmente diferente, pero cuya manifestación principal de la enfermedad es la MCH: uno de los principales genes del sarcómero cardíaco (MYBPC3)25, una enzima implicada en el metabolismo de la globotriacilceramida (GLA), y una enzima implicada en el metabolismo del glucógeno cardíaco (LAMP2)42. El peor pronóstico se observa en los portadores de las mutaciones en LAMP2, que se asocian a la enfermedad de Danon43. La muerte cardiovascular comienza a manifestarse a edades muy tempranas; la mitad de los portadores presentó una muerte cardiovascular a la edad de 40 años, siendo la sobrevida solo de 30% a los 50 años44. Por el contrario, los eventos asociados a mutaciones patogénicas en MYBPC3 se manifiestan más tardíamente, en la mayoría de los casos por encima de los 40 años (aunque hay un 10% de los pacientes que presentaron una muerte cardiovascular antes de esta edad), siendo la mediana de sobrevida de aproximadamente 75 años. Por último, el gen que presentó la menor proporción de eventos cardiovasculares fue GLA (asociado a la enfermedad de Fabry)45-47; las muertes cardiovasculares se manifestaron casi exclusivamente luego de los 40 años, superando el 10% luego de los 60 años (se debe tener en cuenta que una gran proporción de estos pacientes fallecen de falla renal, end-point que no fue evaluado en estas curvas). A la derecha se puede observar la diferencia de presentación entre sexos, que es significativa (p< 0,001%) en los tres casos, aunque con algunas diferencias. En el gen GLA se observa cómo las muertes cardiovasculares comienzan en los varones unos 20 años antes que en las mujeres, manteniéndose la pendiente de las curvas más o menos paralelas (a la edad de 70 años, un 45% de los hombres experimentó una muerte cardiovascular, mientras que este porcentaje fue de 20% en las mujeres). Algo similar sucede con el gen LAMP2, siendo mucho más pronunciada la diferencia entre hombres y mujeres; sin embargo, incluso en las mujeres el pronóstico es muy malo, con una mediana de sobrevida de 50 años48. En este punto es importante remarcar que aunque estas dos son enfermedades con un patrón de herencia ligado a X, las mujeres también desarrollan la enfermedad, generalmente de forma más tardía que los hombres. Por último, las curvas de hombres y mujeres portadores de variantes asociadas a enfermedad en el gen MYBPC3 comienzan a separase luego de los 30 años, manteniendo desde esa edad los hombres una incidencia de muertes cardiovasculares mayor que las mujeres.
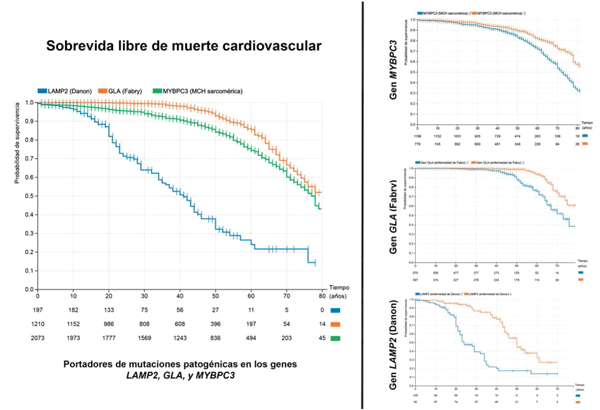
Figura 3: Sobrevida libre de muerte cardiovascular en las mutaciones patogénicas en LAMP2 (azul), GLA (naranja) y MYBPC3 (verde). En la figura de la izquierda se observa la comparación en el global en todos los portadores, mientras que en las figuras de la derecha se compara la sobrevida entre varones (azul) y mujeres (naranja) en cada uno de los genes.
Este caso sirve para ejemplificar la importancia de tener en cuenta en el diagnóstico diferencial la presencia de fenocopias, ya que el pronóstico es totalmente diferente para cada una de estas enfermedades. A veces este diagnóstico diferencial no es tan fácil, especialmente porque son enfermedades muy raras y que no suelen sospecharse14. Por este motivo, todos estos genes asociados a enfermedades consideradas fenocopias deben ser incluidas dentro de los paneles diseñados para cada fenotipo particular.
Esta información que obtenemos de cada gen en particular puede ser una primera aproximación. En muchos genes podemos observar un comportamiento más o menos homogéneo, especialmente de algunas variantes en particular, como las de tipo truncamiento o codón de stop prematuro. Ejemplos de este tipo pueden ser los truncamientos en MYBPC3, LAMP2 o GLA asociados a MCH. Sin embargo, esta aproximación debe ser diferente en otros genes y con otros tipos de variantes.
Pongamos como ejemplo las variantes patogénicas en los tres genes sarcoméricos principales. Si realizamos la gráfica de sobrevida en cada una de ellos (teniendo en cuenta todas las variantes patogénicas en cada gen), obtendríamos la Figura 4. Como puede observarse, la mejor sobrevida se observa en el gen MYBPC3, seguido de las variantes en MYH7, siendo el gen de peor pronóstico TNNT2. Las diferencias son significativas entre las tres curvas (p< 0,001).
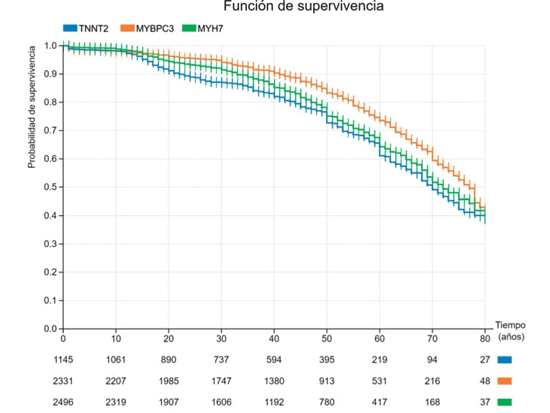
Figura 4: Sobrevida libre de muerte cardiovascular en las mutaciones patogénicas en los tres genes sarcoméricos principales MYBPC3 (naranja), MYH7 (verde) y TNNT2 (azul).
Ahora detengámonos y observemos las curvas de la Figura 5 en donde hemos evaluado todas las variantes de tipo “missense” (cambio de un aminoácido por otro) en el gen MYH7. Si observamos la línea azul, vemos cómo el comportamiento de estas variantes en MYH7 es similar al de todas las variantes patogénicas en MYH7 de la figura anterior, lo que es lógico ya que estas representan más del 95% de las variantes patogénicas descritas en el gen. Si profundizamos más en el análisis, podemos identificar algunas regiones particulares relevantes. Una de ellas es la región conversora (aminoácidos 709-777), que puede asociarse a un peor pronóstico. Incluso dentro de esa región existe un dominio más pequeño (hélice que involucra los residuos 715-722) en donde los portadores empiezan a tener eventos desde la adolescencia, con una caída en la sobrevida que es más pronunciada. En un nivel de detalle aún mayor podemos construir la curva de sobrevida de una variante particular de la que tenemos un número suficiente de portadores que nos permiten establecer un pronóstico fidedigno (p.Arg719Gln). En este análisis podemos ver que la sobrevida libre de muerte cardiovascular a los 50 años, que es 80%-85% en todas las variantes “missense” de MYH7, disminuye a 60% en la región conversora, y es menor de 35% en la hélice o para la variante particular p.Arg719Gln7.
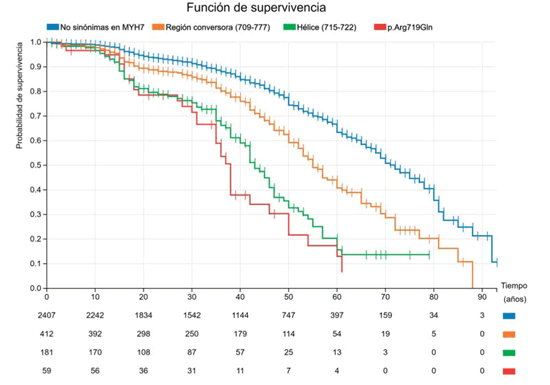
Figura 5: Sobrevida libre de muerte cardiovascular a medida que profundizamos el análisis a un nivel molecular mayor en el gen MYH7: variantes “missense” patogénicas en todo el gen (azul), en la región conversora (naranja), en una hélice de la misma (verde), y finalmente en una mutación en particular (p.Arg719Gln, en rojo). Las diferencias son significativas entre todas las curvas (p< 0,001%) a excepción de la hélice y p.Arg719Gln (p=NS).
Finalmente, un último análisis acerca del pronóstico de algunas variantes en un gen particular. Como veíamos en la figura 4, el gen de la TNNT2 parece ser el que se ha asociado a más muertes cardiovasculares en el seguimiento. Existe la idea de que las mutaciones en este gen se asocian a mal pronóstico, incluso con grados leves de hipertrofia o sin hipertrofia evidente49. La pregunta es si este concepto es válido para todas las variantes en el gen. Para responderla, hemos construido curvas de sobrevida para tres variantes diferentes en TNNT2 que se muestran en la Figura 6. La primera es una variante que tiene un efecto fundador en Galicia (España) y de la que tenemos información de más de 30 familias: p.Asn271Ile50. Se asocia a un fenotipo ligero, en donde es excepcional la presencia de hipertrofia severa. Los eventos son muy raros, prácticamente inexistentes por debajo de los 40 años; los portadores presentan un 90% de sobrevida libre de eventos a los 60 años. Si la comparamos con la segunda variante, p.Arg92Gln, la diferencia es notoria. Esta última se asocia con un fenotipo que suele ser leve a moderado, pero con una incidencia de muertes cardiovasculares (en general dadas por la presencia de muerte súbita) mucho más elevada: los eventos comienzan en la adolescencia, y se mantienen elevados desde entonces, para llegar a un 50% de muertes cardiovasculares a los 60 años51. Por último, una variante de muy mal pronóstico (p.Lys210del), con una sobrevida ligeramente superior a 80% a los 20 años (casi un 20% de los individuos habían sufrido una muerte cardiovascular a esta edad); la sobrevida solo llegaba a 30% a los 60 años. Hay que remarcar que el fenotipo predominante en esta última variante es MCD, y un gran número de las muertes cardiovasculares se deben a fallo cardíaco o trasplante52.
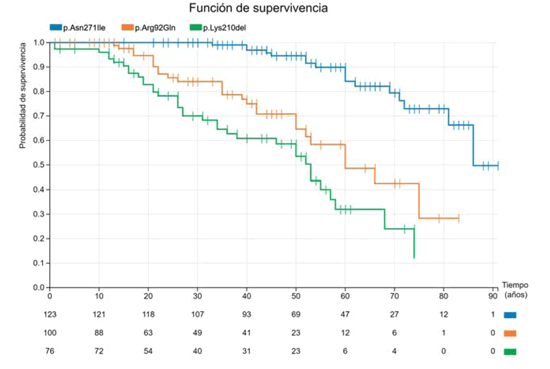
3) Implicaciones terapéuticas
En algunas ocasiones, el resultado del estudio genético puede aportar información importante para decidir un tratamiento. Este valor añadido debe contextualizarse en el cuadro clínico del paciente y nunca interpretarse aisladamente. Como ejemplo, el resultado del estudio genético puede aportar información sobre el momento más adecuado para el implante de un desfibrilador como prevención primaria en MCD. Casi el 50% de las llamadas MCD idiopáticas son de causa genética10. Los sustratos genéticos de este subgrupo pueden ser muy variables y determinar pronósticos diferentes. En la MCD como grupo general, el punto de corte para decidir el implante de desfibrilador como prevención primaria es una fracción de eyección del ventrículo izquierdo igual o menor a 35%. Varios trabajos han demostrado que las mutaciones en el gen de la lámina cardíaca (LMNA) se asocian con un riesgo incrementado de eventos arrítmicos y muerte súbita, que pueden aparecer con deterioros mucho menores de la fracción de eyección53-55. Se ha comparado la sobrevida libre de eventos arrítmicos en portadores de mutaciones en LMNA con respecto a mutaciones en titina (TTN, uno de los genes causales más prevalentes en MCD), y frente a pacientes sin mutación identificada. Los portadores de mutaciones en TTN, junto con los pacientes sin mutaciones, tienen una sobrevida significativamente mejor a los portadores de mutaciones en LMNA56).
Nuestro grupo ha descrito recientemente la asociación de mutaciones de tipo truncamiento en filamina C (FLNC) con el desarrollo de un fenotipo mixto de MCD/MCA de predominio ventricular izquierdo. Estos pacientes también están expuestos a un riesgo mayor de muerte súbita, aun con deterioros ligeros de la fracción de eyección ventricular izquierda, especialmente en presencia de arritmias ventriculares en el Holter y/o fibrosis intramiocárdica detectada mediante resonancia magnética con gadolinio26.
Otro aspecto importante son aquellos subtipos de miocardiopatías que tienen un tratamiento específico que ha demostrado modificar el curso clínico de la enfermedad. Hay dos ejemplos claros secundarios a fenocopias de la MCH. La primera de ellas es la enfermedad de Fabry, que se produce por la acumulación de glicoesfingolípidos como consecuencia de una actividad deficiente de la enzima alfa-galactosidasa A. El tratamiento específico con el aporte parenteral de análogos de la enzima ha demostrado frenar la evolución y disminuir el daño de órgano blanco57. El otro es el caso de la amiloidosis cardíaca; en su variedad por depósito de transtiretina se incluye una forma hereditaria por mutaciones en el gen que codifica esta proteína (TTR). Estudios muy recientes han demostrado que la terapia con tafamidis (un estabilizador específico del tetrámero de transtiretina) produce una disminución en la mortalidad cardiovascular y hospitalizaciones por insuficiencia cardíaca en pacientes con amiloidosis TTR58. En ambas patologías, el estudio genético puede ser una herramienta importante en su diagnóstico diferencial inicial.
Limitaciones del estudio genético en miocardiopatías
El análisis genético debe ser considerado una prueba complementaria más en el abordaje de las miocardiopatías. Su utilización de ninguna manera reemplaza una correcta evaluación clínica. Los resultados genéticos deben integrarse con los de otras pruebas complementarias (electrocardiograma ECG), ecocardiograma, Holter ECG, prueba de esfuerzo, cardiorresonancia, etc.) para construir una panorámica más completa de la patología que afecta al paciente y su familia. Es una equivocación pensar que el estudio genético por sí solo va a resolver todas las incertidumbres diagnósticas y pronósticas. En un porcentaje variable de pacientes con diagnóstico clínico inequívoco, el estudio genético puede no identificar la causa de la enfermedad. El avance continuo de las tecnologías y el conocimiento están reduciendo la cantidad de estudios genéticos negativos o no informativos y aumentando su rentabilidad.

