











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
La miocardiopatía arritmogénica del ventrículo derecho (MCAVD) fue reconocida como una entidad específica en la década de 1970 cuando pacientes con taquicardia del ventrículo derecho (VD) resistente a los medicamentos se sometieron a cirugía guiada por un mapeo epicárdico. Se demostró su origen en el VD con dilatación del mismo, extensión hacia el ventrículo izquierdo (VI) e infiltración linfovascular en muestras de biopsia. Fue inicialmente denominada displasia, dado que se otorgaba un papel principal al trastorno del desarrollo de la pared del VD. Posteriormente, la evidencia de estudios morfológicos y clínicos mostró que no se trataba de un defecto estructural presente en el nacimiento (como la anomalía Uhl), sino de una enfermedad adquirida y progresiva del miocardio. El término displasia fue reemplazado y la afección se conoció como “miocardiopatía del VD”. Durante el mismo período, las arritmias ventriculares se reconocieron como la presentación clínica predominante de la enfermedad, y muchos informes se refirieron a ella como “displasia arritmogénica del VD” o “miocardiopatía arritmogénica del VD”. A pesar de esta evolución en el pensamiento y cualquiera sea su denominación, la MCAVD ha sido incluida en la clasificación del grupo europeo de miocardiopatías desde 19941).
Definida como una patología cardíaca de origen genético, cuya base molecular radica en alteraciones a nivel de los desmosomas, su hallazgo morfológico más constante es la sustitución progresiva de miocitos del VD por tejido graso, en forma difusa o segmentaria desde el epicardio hasta el endocardio. Se crea así un sustrato anatómico para el desarrollo de arritmias ventriculares y disfunción. Las descripciones iniciales se centraban en la base anatómica arrítmica de ciertas zonas del VD (el llamado “triángulo de la displasia”). Actualmente el espectro se ha ampliado reconociendo manifestaciones difusas en el VD, afectación única del VI (más frecuente en subepicardio de pared posterolateral) y alteración biventricular en fase dilatada, a menudo indistinguible de la miocardiopatía dilatada (MCD).
Los estudios poblacionales han demostrado una incidencia de 1,01 por 100.000 habitantes al año entre los 15 a 19 años de edad, siendo muy rara su aparición antes de los 10 años2. La prevalencia es mayor en hombres que en mujeres (2,4:1)3, lo que puede explicarse por la influencia directa de las hormonas sexuales en la expresión fenotípica de la enfermedad o por las diferencias basadas en el sexo en la práctica o intensidad del ejercicio4. En la región italiana del Véneto se estima una prevalencia de un caso por 1.000 o 10.000 personas; siendo responsable del 20% de las muertes súbitas en jóvenes y la causa más frecuente de muerte súbita en atletas italianos5,6. Se ha reportado que aproximadamente el 50% de los pacientes afectados tienen antecedentes familiares positivos.
El diagnóstico de la MCAVD es probablemente el más desafiante en el campo de las miocardiopatías hereditarias debido a la ausencia de criterios diagnósticos únicos específicos, su expresividad variable y su penetrancia incompleta en los familiares de los afectados. A esto debe adicionarse su pobre expresión clínica en la edad pediátrica, aun en aquellos con estudio genético positivo, por lo que deben reconocerse elementos de sospecha para orientar y focalizar el estudio especializado.
Genética molecular
La MCAVD es causada por mutaciones de los genes que codifican las proteínas estructurales y de adhesión de los miocitos, específicamente a nivel de los desmosomas (Figura 1), con pérdida del contacto intercelular normal. Se han encontrado mutaciones en cinco proteínas desmosómicas en aproximadamente el 60% de los pacientes7,8, y aproximadamente el 50% de los casos presenta un patrón de herencia autosómico dominante. La mutación más común es la que afecta la placofilina-2 (PKP2), que se ha aislado hasta en el 46% de casos índice y en 61% de familiares de afectados9. Otras proteínas desmosómicas que se ven afectadas incluyen desmoplaquina y desmogleína (las cuales, junto a la PKP2, dan cuenta del 90% de las mutaciones totales encontradas) y en menor frecuencia desmocolina y placoglobina. Se han descrito dos formas autosómicas recesivas de MCAVD debido a mutaciones en la placoglobina, que causa la enfermedad de Naxos, y en la desmoplaquina, que causa la enfermedad de Carvajal, que afecta predominantemente al VI. Estos trastornos también implican anormalidades en las proteínas desmosomales en la piel y el cabello. Aproximadamente del 20% al 30% de los pacientes con MCAVD tienen antecedentes familiares de ésta o de muerte súbita. Una vez que una persona tiene una mutación, ya sea heredada o de novo, hay un 50% de posibilidades de que ésta se transmita a los hijos de la afectada.
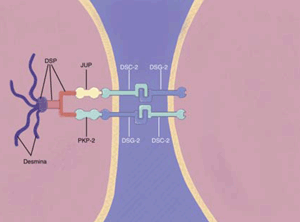
Figura 1: Genética molecular de la MCAVD. Se han descrito tres clases de proteínas desmosómicas: 1) Cadherinas, que incluyen la desmolecina (DSG-2), y la desmocolina (DSC-2), que se extienden por la membrana celular para conectar los miocitos. 2) Plaquinas, que incluyen la desmoplaquina (DSP), que ancla los desmosomas a los filamentos intermedios. 3) Proteínas del armadillo, incluyen placoglobina (JUP) y placofilina (PKP-2), que conectan la desmoplaquina y las cadherinas.
El deterioro de la función del disco intercalar da lugar al desprendimiento de los miocitos y a su muerte, sobre todo en situaciones de estrés estructural. Esta hipótesis patogénica explicaría por qué la MCAVD se asocia principalmente a las áreas más delgadas de la pared del VD y posterolateral del VI, que son estructuralmente más vulnerables al estrés mecánico.
Se ha demostrado una significativa remodelación de los discos intercalares miocitarios en estos pacientes, caracterizada por una marcada disminución del número de desmosomas y por la pérdida de la integridad estructural de los mismos10.
Resultados recientes han demostrado que la MCAVD tiene en realidad una base genética más compleja, lo que ayudaría a entender la penetrancia incompleta y la expresión fenotípica variable, incluso entre miembros de la misma familia y con la misma mutación. Asimismo, un porcentaje cercano al 10% de los pacientes pueden tener más de una mutación en el mismo o diferentes genes asociados al desarrollo de la miocardiopatía, e incluso en algunos casos es necesaria la presencia de dos o más mutaciones en varios genes (mutación digénica) para el desarrollo de la patología11.
Interesantemente, en comparación con los pacientes con una sola mutación asociada a la miocardiopatía, los portadores de mutaciones múltiples muestran manifestaciones más graves de la enfermedad, con mayor prevalencia de afectación del VI, mayor dilatación del VD, elevado riesgo de eventos arrítmicos graves, taquicardias ventriculares (TV), síncopes y mayor frecuencia de antecedentes familiares de muerte súbita12.
Una persona que tiene una mutación para la MCAVD hereda el riesgo de tener la enfermedad pero puede o no desarrollar signos y síntomas de ésta; característica que se repite en las miocardiopatías genéticas donde encontramos una gran variabilidad clínica ante genotipos similares, desde la ausencia de síntomas hasta insuficiencia cardíaca o debut con muerte súbita. Esta heterogeneidad sugiere que otros factores juegan un rol importante en la modificación del fenotipo clínico, ya sea para la potenciación o la protección de la enfermedad. Se ha hipotetizado sobre la existencia de algunos de ellos, como genes moduladores, polimorfismo, otros genes aún desconocidos, factores del medio ambiente y endógenos (edad, sexo, ejercicio físico, drogas, hormonas, infecciones virales y estrés emocional)13).
Criterios diagnósticos en pediatría
Habitualmente la MCAVD se diagnostica entre los 20 y 40 años de edad. Su diagnóstico es de difícil sospecha en adolescentes y la presentación de características clásicas del electrocardiograma (ECG) y de la imagenología complementaria es excepcional en los menores de 10 años. El total de niños afectados por MCAVD en las series clínicas no sobrepasa el 5% al 20% del total de enfermos, y generalmente se encuentran entre los 15 a 18 años14. Desde el punto de vista clínico se reconocen cuatro etapas:
1. Fase temprana o silente (que abarca gran parte de la edad pediátrica), característicamente asintomática, pero que hasta en 20% de los casos debuta con muerte súbita.
2. Fase inestable, en la que predominan arritmias sintomáticas presumiblemente originadas en el VD.
3. Falla del VD, con conservación relativa de la función del VI.
4. Dilatación biventricular progresiva, asociada a embolias y fibrilación auricular.
Se puede reconocer entonces que tanto la presentación clínica como el curso de la MCAVD están determinados por los eventos arrítmicos, y la posterior alteración sistólica biventricular que conduce a insuficiencia cardíaca. En este contexto, resulta fundamental dilucidar en qué niño con historia inespecífica de palpitaciones o síncope existen elementos clínicos o electrocardiográficos que permitan predecir el posterior desarrollo de la miocardiopatía. Lamentablemente, aún faltan estudios para poder recomendar una conducta9. Las guías actuales (que datan del año 2010) han permitido un aumento de la sensibilidad en el diagnóstico de la MCAVD, determinado principalmente por la incorporación del screening genético y la mayor sensibilidad para detectar disfunción y dilatación del VD a través de la resonancia magnética15.
Es posible reconocer los siguientes escenarios de presentación clínica en pediatría:
1. Paciente asintomático familiar de un probando, con o sin genotipo asociado a patogenicidad: probablemente sea el grupo más frecuente con el transcurso de los años debido a la asociación de la miocardiopatía con el hallazgo de mutaciones. Al respecto, el diagnóstico clínico de la MCAVD en un individuo modifica el riesgo genético de sus familiares de primer grado, que pasa de 1:1.000-1:5.000 de la población general a 1:2. Los criterios definidos en el año 2010 para diagnóstico de la MCAVD lo reconocen al considerar que la presencia inequívoca de enfermedad en un familiar de primer grado constituye un criterio mayor (siendo en el año 1994 un criterio menor)19. Si el diagnóstico clínico de MCAVD se da en un familiar de segundo grado o hay antecedentes de MCAVD en un familiar de primer grado, pero no se puede determinar si dicho familiar cumple los criterios de laTask Force, se considera un criterio menor.
2. Paciente con ECG alterado solicitado por screening precompetitivo: pueden identificarse elementos sugerentes que motivan la profundización y complemento con estudios imagenológicos. La enfermedad debe sospecharse incluso en individuos asintomáticos sobre la base de estas anomalías, siendo la más frecuente la inversión de la onda T en las derivaciones precordiales derechas, sin que haya un bloqueo de rama derecha del haz (BRDH) completo. La prevalencia de la inversión de la onda T en las derivaciones precordiales derechas en sujetos sanos de más de 14 años de edad se observa solo en 4% de las mujeres y en 1% en los varones sanos16. Por consiguiente, este hallazgo se considera una anomalía mayor para los criterios diagnósticos actuales.
3. Palpitaciones: asociadas a la presencia de arritmias ventriculares. Las arritmias ventriculares a menudo constituyen la forma de presentación inicial de la enfermedad en adolescentes y adultos jóvenes. La TV sostenida o no sostenida y las extrasístoles ventriculares tienen habitualmente una morfología de bloqueo de rama izquierda (lo cual refleja su origen en el VD), asociado a un eje superior del QRS (QRS negativo o indeterminado en las derivaciones II, III y aVF y positivo en aVL), considerado actualmente un criterio mayor. Al respecto, la presencia de más de 500 extrasístoles ventriculares en un registro de Holter de 24 horas constituye un criterio menor.
4. Síncope y muerte súbita: en especial en aquellos con presentación atípica, por ejemplo asociados al esfuerzo físico o que ocurren en decúbito; el síncope que no refiere pródromos o aquel con historia familiar de muerte súbita, lo cual debe obligar a descartar arritmias ventriculares. La muerte súbita se asocia al esfuerzo, y puede ser la primera manifestación, en especial en jóvenes deportistas17. De acuerdo a lo reportado en el estudio de la región de Véneto, Italia, los adolescentes y adultos jóvenes que participan en actividades deportivas tienen 2,8 veces mayor riesgo de muerte cardiovascular súbita que sus contrapartes no atléticas (análisis prospectivo de muerte súbita en jóvenes)18.
En 2010 un grupo de trabajo internacional revisó las recomendaciones diagnósticas que databan de 199419. En ellas se adicionó el estudio genético molecular como criterio mayor y se incorporaron criterios cuantitativos para diagnosticar anomalías imagenológicas del VD (tanto en ecocardiograma como en resonancia magnética). Esto permite su clasificación en categorías diagnósticas: “definitivo”, “límite” o “posible” de MCAVD (Tabla 1).
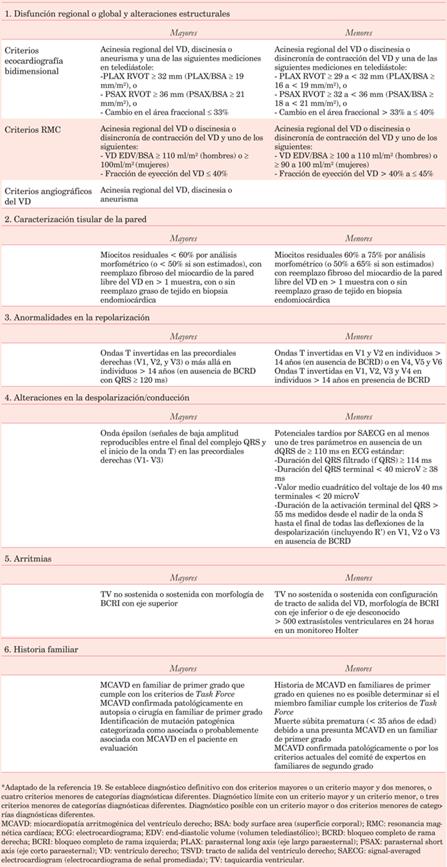
A pesar de lo anterior, y considerando que los estudios posteriores han demostrado un aumento en la sensibilidad para definir los casos definitivos y probables (tanto en adultos como en adolescentes), los seguimientos han mostrado dos inconvenientes. En primer lugar, el diagnóstico sigue siendo problemático debido a la baja especificidad de las anomalías electrocardiográficas, las causas múltiples de arritmias con origen en el VD, la dificultad en la disponibilidad de diferentes modalidades de imagen y los resultados a veces poco categóricos de las pruebas genéticas. En segundo lugar, el grupo con menor representación en las cohortes sobre las que se basan las recomendaciones es el de los menores de 15 años, lo que dificulta la extrapolación de sus conclusiones.
Estudios complementarios
- Electrocardiograma: tanto las extrasístoles ventriculares como las TV muestran complejos QRS positivos en DI, eje eléctrico variable e imagen de bloqueo de rama izquierda que orienta al origen en el piso o en la punta del VD. En ocasiones, se observa un complejo QRS de predominio negativo en DI, con eje eléctrico normal o desviado a derecha, lo cual debe hacer sospechar en que el inicio se localiza en el infundíbulo pulmonar. Se ha observado la presencia de arritmias auriculares (extrasístoles auriculares, taquicardia, fibrilación o flutter auricular) en 20% a 25% de los pacientes jóvenes como indicio de la enfermedad. En la MCAVD las alteraciones que se pesquisan clásicamente en ritmo sinusal son las siguientes (Figura 2):
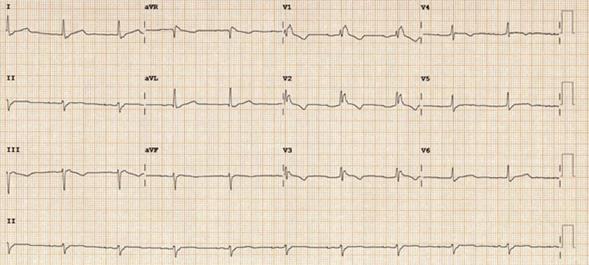
Figura 2: Electrocardiograma de paciente con diagnóstico de MCAVD. Se observa QRS ensanchado y ondas T (-) desde V1 hasta V4.
- Onda T negativa más allá de V1 (en relación con el grado de compromiso y dilatación del VD), este criterio es válido para los mayores de 14 años.
- QRS ensanchado, especialmente en V1-V3 (implica grados variables de demora en la activación del VD, desde bloqueo incompleto hasta bloqueo de alto grado, posiblemente por alteración periférica a nivel del miocardio contráctil y no en las ramas del haz de His).
- Incremento de onda S> 55 ms en V1-V3.
- Onda épsilon: presente en alrededor de un 30% de los casos. Representa potenciales eléctricos de baja amplitud que aparecen al final del complejo QRS y al inicio del segmento ST. Visibles en precordiales derechas, corresponden a áreas de activación retardada de algunas fibras miocárdicas, por sustitución fibrosa o fibroadiposa en el VD.
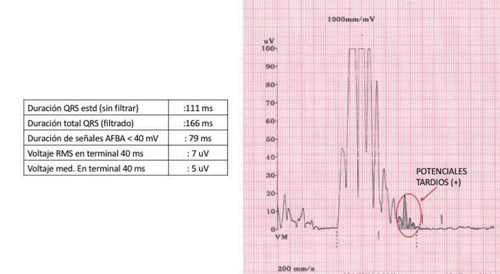
- Electrocardiograma de señales promediadas: fue empleado por primera vez para la investigación de potenciales tardíos en los pacientes que durante cirugías cardíacas exhibían potenciales retrasados en los registros epicárdicos. El hallazgo de potenciales tardíos (examen positivo) mediante el ECG de señales promediadas corresponde a áreas de conducción eléctrica lenta en el miocardio afectado, potencial sustrato de arritmias ventriculares de reentrada bajo ciertas circunstancias electrofisiológicas (Figura 3). Se cree, además, que pueden ser marcadores de la forma difusa de compromiso del VD, ya que la magnitud de la positividad guarda relación con el volumen final diastólico del VI. Los potenciales tardíos son uno de los marcadores más sensibles de esta miocardiopatía (85% en algunas series), por lo que se solicitan en los casos en los que existe sospecha clínica o electrocardiográfica cuando aún no se han objetivado arritmias. En niños, los valores obtenidos deben ser considerados en relación con la edad del paciente, descritos en la literatura20.
- Radiografía de tórax: se puede observar cardiomegalia moderada. Sin embargo, lo habitual en pediatría es que exista una leve dilatación del VD, por lo que generalmente el índice cardiotorácico es < 0,6.
- Ecocardiograma Doppler color: lo más sugerente de esta patología en niños es la dilatación exclusiva de las cavidades cardíacas derechas (en particular del VD). Otros hallazgos que deben hacer sospechar son: adelgazamiento parietal, discinesia de la pared inferobasal y apical, cambios estructurales de la banda moderadora (engrosamiento e hiperrefringencia), desorganización trabecular, dilatación aislada del tracto de salida del VD y saculaciones en la pared libre del VD formando aneurismas localizados.
- Resonancia magnética cardíaca (RM): durante la última década, la RM ha emergido como la modalidad de imagen de elección en la MCAVD, pues permite una evaluación morfológica y funcional no invasiva, así como la caracterización tisular en un solo procedimiento. Ha demostrado ser más sensible que la ecocardiografía para detectar la dilatación ventricular temprana y la disfunción en niños15, incluso en aquellos con genotipo positivo. El papel de la RM en el diagnóstico de la MCAVD ha sido reconocido en los criterios del 2010, incorporando valores precisos para la categorización de enfermos, lo cual ha redundado en un aumento del valor predictivo positivo de estas recomendaciones (desde 23% en los criterios originales a 55% en los criterios revisados)21. Las anormalidades del VD descritas en la MCAVD son numerosas e incluyen: reducción global de la función del VD y crecimiento de este, alteraciones regionales y finas con terminología variable (abultamientos focales, microaneurismas, dilatación segmentaria, hipocinesia regional, etcétera). Se debe considerar en particular la importancia de las alteraciones de la motilidad parietal regional en la región subtricuspídea, como el “signo del acordeón”22, que se produce por una disincronía de la contracción en una región de miocardio focalizada. Aunque el realce tardío con gadolinio (LGE) es una técnica validada para objetivar la fibrosis miocárdica, no ha sido incorporado en los criterios diagnósticos para la identificación del reemplazo fibroadiposo característico de la MCAVD. Como limitantes cabe destacar el adelgazamiento del VD que puede dificultar la detección del LGE, además de que puede tratarse de un hallazgo poco específico ya que se ha descrito en otras situaciones clínicas como la sarcoidosis, miocarditis, amiloidosis y la miocardiopatía dilatada. A pesar de estas limitaciones, el LGE se ha observado en hasta 88% de los pacientes23. El LGE también puede ser útil en el manejo de pacientes con MCAVD. Algunos estudios han demostrado una adecuada correlación entre el LGE del VD, la histopatología y las arritmias ventriculares inducibles en el estudio electrofisiológico24. De ahí que la identificación de LGE por RM puede proveer una guía para los estudios electrofisiológicos y para la realización de biopsias endomiocárdicas. Siempre se debe considerar (en particular para los pacientes pediátricos) que las anormalidades eléctricas preceden a los cambios estructurales detectados por RM. En conclusión, la evaluación de MCAVD no se debe basar en una prueba aislada, sino en un complemento de los exámenes señalados.
- Estudio electrofisiológico y ablación con radiofrecuencia: los hallazgos electrofisiológicos sugieren que el mecanismo de la TV de esta enfermedad es por reentrada. En las áreas anormales es frecuente observar un aumento del umbral de estimulación. Esto hace necesario múltiples intentos de estimulación ventricular programada y el uso de salvas rápidas, junto a la infusión endovenosa continua de isoproterenol. Elevar la frecuencia cardíaca a 100-120 latidos por minuto facilita la inducción de la arritmia ventricular (Figura 4).
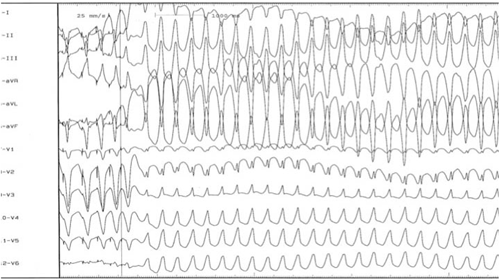
Papel del test genético en la miocardiopatía arritmogénica del ventrículo derecho
La identificación de la mutación genética causante de la enfermedad en un paciente con MCAVD permite:
- Identificar a los miembros de la familia genéticamente afectada en una fase preclínica y descartar como enfermos a las personas no portadoras.
- Estratificar el riesgo de muerte súbita.
- Confirmar el diagnóstico en un individuo afectado con sospecha clínica.
- Distinguir la MCAVD de otras patologías fenotípicamente similares (fenocopias) como la miocardiopatía dilatada con compromiso biventricular, la miocardiopatía inflamatoria y la afectación cardíaca en distrofias musculares.
Es importante recordar que las pruebas genéticas moleculares solo pueden apoyar un diagnóstico evidente o una sospecha clínica, pero no hacen el diagnóstico de MCAVD en forma aislada. De hecho, los portadores de mutaciones pueden no tener el fenotipo (penetrancia incompleta) o presentar diferentes grados de manifestaciones clínicas (expresión variable). Así, un resultado genético positivo solo puede ser parte de un enfoque clínico en el que se combinen múltiples fuentes de información diagnóstica como cambios en el ECG, arritmias ventriculares y cambios morfofuncionales del VD.
Sin duda, el principal impacto clínico es la posibilidad de estudiar las familias con un individuo afectado (probando), aun cuando estos integrantes no hayan demostrado signos o síntomas de la enfermedad, lo cual es especialmente válido en pediatría pues los síntomas generalmente comienzan recién en la adolescencia, luego de una larga fase preclínica.
La evaluación inicial de los miembros de la familia debe efectuarse si el probando ha sido diagnosticado con una mutación desmosomal, caso en el cual se debe realizar un cribado en cascada de la familia incluidos los niños. Por el contrario, la probabilidad de diagnosticar MCAVD en un miembro de la familia de un probando, sin una mutación genética, es mucho menor9. Del mismo modo, la probabilidad de arritmias ventriculares sostenidas o muerte súbita cardíaca en el miembro de la familia de un probando, sin una mutación genética conocida, es muy reducida. Los individuos no portadores (que en series representan hasta el 50% de los evaluados) en la práctica pueden considerarse como saludables y pueden estar seguros de la no transmisión de la enfermedad a sus hijos.
Para los familiares que tienen la mutación genética del probando, o pertenecen a una familia con un caso sin mutación genética conocida, la evaluación cardíaca, cuando no existe evidencia de la enfermedad, debe comenzar entre los 10 a 12 años de edad (dado que la enfermedad raramente se expresa antes de esa fecha). Las pruebas sugeridas incluyen: ECG, ecocardiograma Doppler color y Holter. Se puede complementar con test de esfuerzo. En caso de encontrarse alguna anomalía sugerente, se recomienda efectuar una RM.
Se sugiere que la evaluación se haga anualmente hasta los 25 años, y si es negativa, cada tres años hasta aproximadamente los 40-50 años, y luego cada cinco años. El seguimiento se puede detener entre los 50 a 60 años dado que es poco común su expresión luego de esta edad.
El diagnóstico precoz de la MCAVD mediante análisis genético molecular se justifica porque permite focalizar el seguimiento en los individuos de riesgo y promover una estrategia preventiva específica en base a modificaciones del estilo de vida, como desaconsejar la participación en actividades deportivas de alta competición. Permite además realizar un seguimiento clínico dirigido a la identificación de síntomas de alarma (como variaciones ecocardiográficas o electrocardiográficas, o ambas, o arritmias ventriculares), y un tratamiento profiláctico adecuado con betabloqueadores, amiodarona o incluso con un desfibrilador automático implantable (DAI).
Para el caso particular de la MCAVD, los resultados de la prueba genética deben interpretarse con cautela. Un hallazgo positivo indica que una mutación causante de la enfermedad se identificó en ese individuo; una variante de significado clínico desconocido indica que el rol patogénico de la enfermedad no puede establecerse claramente y una prueba negativa podría significar que el paciente no va a desarrollar la enfermedad, o bien que tiene una mutación en una región no analizada del gen o en otro que no forma parte del estudio genético.
Estratificación de riesgo en pediatría
A pesar de que estudios en los años 9025 demostraron que la sensibilidad de los criterios diagnósticos originales de MCAVD es similar en población joven y adulta, el aislamiento de mutaciones genéticas asociadas al desarrollo del fenotipo de la enfermedad ha motivado un cambio en el enfoque de la detección temprana de esta patología en los pacientes pediátricos. Lo anterior no hace sino corroborar la importancia de efectuar un estudio en cascada en la familia de un probando (que probablemente sea un paciente adulto) para así reconocer al paciente pediátrico potencial de desarrollar la enfermedad.
La estratificación de riesgo se ha logrado establecer en adultos. Su aplicabilidad en pediatría (en especial las cifras referidas a los eventos por año) puede verse limitada, dado que las recomendaciones del año 2010 se basan en el Estudio Multidisciplinario de América del Norte de Displasia del Ventrículo Derecho, que incluyó 108 personas, de las cuales sólo nueve tenían entre 12 y 18 años26.
Un hallazgo a destacar es que las anomalías eléctricas parecen manifestarse antes que las anomalías estructurales. Así, en pacientes asintomáticos portadores de mutaciones conocidas, es más frecuente encontrar anomalías en exámenes previos al evento de presentación clínica (hasta 61% de pacientes en ECG o Holter) que pesquisar anormalidades estructurales en la RM27. Parece razonable, por lo tanto, que la evaluación del niño debe enfocarse en una vigilancia cercana de la expresión de anormalidades eléctricas. A pesar de lo anterior, las anormalidades estructurales se encuentran presentes especialmente cuando se estudian con RM pacientes sospechosos (por clínica o por genotipo sugerente), lo cual ha permitido aumentar el número de pacientes con enfermedad definida de acuerdo a los criterios establecidos en 201028. A diferencia de la población adulta, en donde puede evidenciarse la fibrosis o aneurismas de VD, son las anomalías regionales de movimiento de la pared lo más frecuente en pacientes jóvenes (hasta en 83% de los pacientes con enfermedad definida y 53% con enfermedad probable)15.
La relación entre el tipo de mutación genética y el curso clínico es un área de estudio actual. La penetrancia de la enfermedad es mayor en pacientes portadores de genotipos complejos, ya sea pacientes con heterocigosidad compuesta (un solo gen con más de una mutación que causa la enfermedad) o aquellos con heterocigosidad digénica (dos genes diferentes con mutaciones patogénicas). Junto al sexo masculino, recientemente este patrón genético ha sido descrito como predictor independiente de muerte súbita cardíaca o TV. Así, en teoría, en este grupo de niños el seguimiento clínico e imagenológico seriado debiera estar indicado como parte de su evaluación.
Las series de pacientes adultos han mostrado que los principales factores de riesgo incluyen: disfunción o dilatación ventricular derecha, disfunción izquierda (generalmente no presente en la edad pediátrica) y la TV espontánea (pesquisada en los monitoreos ambulatorios o en el test de esfuerzo). Las arritmias ventriculares y el síncope de causa no clara deberían ser los elementos de sospecha en la población pediátrica, y motivar el estudio genético oportuno en presencia de historia familiar compatible o fenotipo electrocardiográfico y/o ecocardiográfico. Se han identificado varios factores de riesgo menores pero su asociación con resultados desfavorables se basa en estudios limitados y con resultados contradictorios29. Aunque las pruebas electrofisiológicas intracardíacas se han utilizado tradicionalmente para evaluar el riesgo de arritmias ventriculares, el valor pronóstico de la TV y de la fibrilación ventricular inducida en los estudios electrofisiológicos en pacientes con MCAVD asintomática sigue sin estar claro30.
Consideraciones en el manejo de la miocardiopatía arritmogénica del ventrículo derecho
El rol del ejercicio en su desarrollo
Numerosos estudios han establecido una relación entre el ejercicio y la expresión y progresión de la enfermedad. Existe consenso en que el deporte per se no es una causa del aumento de la mortalidad, sino que actúa como desencadenante del paro cardíaco en presencia de una enfermedad cardiovascular subyacente. Se cree que la adhesión deteriorada célula a célula por los desmosomas genéticamente alterados aumenta el estiramiento de la pared del VD cuando se somete a ejercicio, con el consiguiente aumento de la tensión parietal. El estrés parietal del VD en el ejercicio máximo en los atletas aumenta 170% en comparación con solo un aumento de 23% en la pared del VI. El VD de individuos con mutaciones desmosómicas puede ser particularmente vulnerable a la remodelación patológica en respuesta al ejercicio. En estos pacientes, el ejercicio de resistencia lleva a un estiramiento anormal de la pared del VD y puede así facilitar el desacoplamiento de los miocitos, lo que conduce a inflamación, fibrosis, adipocitosis y deterioro directo del acoplamiento eléctrico, que predispone a las arritmias ventriculares18. Uno de los primeros en destacar este importante vínculo fue el estudio de muerte súbita cardíaca en la región italiana de Véneto, que encontró que los atletas jóvenes tenían un riesgo cinco veces mayor de morir de MCAVD en comparación con los no atletas21, lo cual fue corroborado en cohortes de pacientes con mutaciones desmosómicas4, donde se asoció el ejercicio de resistencia a una mayor probabilidad de cumplir con los criterios de diagnóstico del 2010 para MCAVD y al desarrollo de TV, fibrilación ventricular o insuficiencia cardíaca. Los atletas de resistencia también desarrollan síntomas a una edad más temprana. Interesantemente en este grupo, disminuir la duración del ejercicio reduce el riesgo arrítmico.
A pesar de lo anterior, hay evidencia que sugiere que existe un nivel de ejercicio seguro. En el estudio multidisciplinario de Estados Unidos, los deportes competitivos se asociaron con una duplicación del riesgo de taquiarritmias ventriculares y muerte30, mientras que el riesgo fue similar en un informe reciente del registro Johns Hopkins, que encontró que los miembros de la familia con mutaciones desmosómicas tenían una baja incidencia de ser catalogados como enfermos si limitaban el ejercicio a menos de 750 minutos/MET/semana31, que representa el menor nivel de ejercicio recomendado por la American Heart Association en prevención primaria.
Actualmente, las recomendaciones de ejercicio tanto en Estados Unidos como en Europa son bastante restrictivas para los pacientes con MCAVD. Las directrices de la 36ª Conferencia de Bethesda recomiendan que a los pacientes con MCAVD, definida o probable, se les debe restringir la participación en deportes competitivos con la excepción de aquellos clase I-A (baja intensidad)33. Otros grupos incluyen limitaciones para portadores de mutaciones asintomáticos. Si bien aún no se ha determinado si estas restricciones se pueden individualizar, parece adecuado que dada la variabilidad fenotípica se aconseje la restricción moderada para todos los pacientes y se reserve la restricción severa solo para aquellos con enfermedad progresiva. En este sentido, la evidencia disponible actualmente indica que los miembros de la familia con un fenotipo negativo (portadores genéticos sanos o aquellos con un genotipo desconocido) no necesitan ningún tratamiento específico que no sea la restricción deportiva; sin embargo, la evaluación clínica de por vida con el uso de pruebas no invasivas al menos cada dos años, está justificada debido a la penetrancia relacionada con la edad y la naturaleza progresiva de la MCAVD.
Si bien la prescripción de ejercicio de este tipo puede ser difícil de aplicar en adolescentes, es importante abordar los hábitos de vida con los pacientes regularmente durante el seguimiento.
Opciones terapéuticas
Los objetivos del tratamiento clínico de la MCAVD en pediatría incluyen prevenir o retrasar la progresión de la enfermedad clínica. Una vez establecida esta, se deben implementar medidas para reducir el riesgo de muerte súbita cardíaca y mejorar la calidad de vida al aliviar los síntomas arrítmicos y de insuficiencia cardíaca. Lamentablemente, los enfoques terapéuticos actuales de la MCAVD son paliativos y alivian parcialmente los síntomas y el riesgo de muerte cardíaca súbita, pero no impiden el desarrollo o la progresión del proceso de la enfermedad.
Un tratamiento curativo definitivo requerirá necesariamente de un conocimiento más profundo de los mecanismos biológicos y de los factores ambientales involucrados en la patogénesis de la enfermedad.
El DAI ha demostrado tener un papel importante para la prevención de la muerte súbita cardíaca, con una disminución de la incidencia desde un 16% en pacientes adultos sin DAI a 0,6% en aquellos con el dispositivo, en seguimientos de cohortes. Aún más, en seguimientos de familiares (con edad promedio de 36 ± 14 años) que cumplen criterios para MCAVD, también se demostró el beneficio del DAI, y solo se reportó muerte súbita cardíaca en aquellos que no tenían un dispositivo9.
Faltan estudios acerca del beneficio del DAI para la prevención primaria de muerte súbita cardíaca en la población pediátrica, aunque su ocurrencia ha sido descrita en individuos menores de 18 años, como lo demuestra el seguimiento efectuado en Ontario, donde de 116 niños con muerte súbita cardíaca, 14 pacientes cumplieron criterios para MCAVD definida o probable y cinco de estas muertes ocurrieron durante una actividad física moderada o vigorosa2).
La decisión de implantar un DAI en un adolescente debe considerar el riesgo de muerte súbita cardíaca en forma individual, y este es más alto en pacientes que ya han tenido TV sostenida o en los que presentan ectopía ventricular frecuente y taquiarritmias ventriculares no sostenidas. Faltan estudios de seguimiento para poder proponer una conducta terapéutica basada en la estratificación del riesgo como la propuesta en adultos , si bien esta es adaptada para la población pediátrica en la práctica clínica habitual (Figura 5). Considerando lo anterior, en pacientes asintomáticos (como la mayoría de la población pediátrica), sin factores de riesgo y en portadores de genes patogénicos, generalmente no hay indicaciones para la implantación preventiva de un desfibrilador debido al bajo riesgo de arritmias y al riesgo significativo de complicaciones relacionadas con los dispositivos y los electrodos durante el seguimiento a largo plazo (tasa estimada de 3,7% por año).
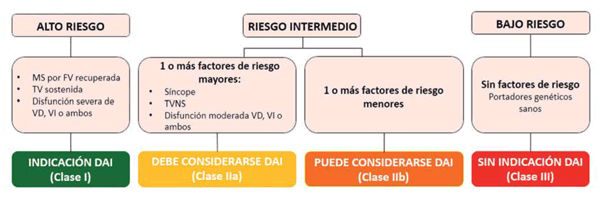
Figura 5: Los grupos de riesgo se han definido sobre la base de la probabilidad estimada de un evento arrítmico mayor (muerte súbita cardíaca, paro cardíaco por fibrilación ventricular, taquicardia ventricular sostenida o un evento que requiera intervención con desfibrilador) durante el seguimiento, en relación con eventos arrítmicos previos o factores de riesgo. Un riesgo anual estimado de más de 10% define el grupo de alto riesgo; entre 1% y 10% el grupo de riesgo intermedio, y un riesgo por debajo de 1% al de bajo riesgo.
Los betabloqueadores tienen un beneficio demostrado en prevenir la muerte súbita cardíaca y mejorar la supervivencia en pacientes con insuficiencia cardíaca. Así, es razonable esperar que estos fármacos tengan un efecto similar en los pacientes con MCAVD, ya que las muertes por arritmias ventriculares son más frecuentes durante el ejercicio. En aquellos con afectación clínica, actualmente los betabloqueadores se recomiendan tanto para la prevención de arritmias como para la reducción del estrés de la pared ventricular derecha. En pacientes con arritmias ventriculares, la terapia con fármacos antiarrítmicos ofrece la posibilidad de mejorar los síntomas, aunque no hay pruebas de que confiera protección contra la muerte súbita cardíaca. La amiodarona, sola o en asociación con los bloqueadores beta, y el sotalol son los fármacos más efectivos, ya que combinan los efectos sinérgicos de las propiedades antiarrítmicas de clase III y el bloqueo betaadrenérgico.
La ablación con catéter de radiofrecuencia es una opción terapéutica para los pacientes que tienen episodios de TV monomórfica sostenida. Sin embargo, debe considerarse como un abordaje paliativo debido a la alta frecuencia de recurrencias y a que la ablación no ha demostrado eficacia para prevenir la muerte súbita cardíaca. El pobre resultado a largo plazo se ha atribuido a la naturaleza progresiva de la MCAVD, que conduce al desarrollo de múltiples focos arritmogénicos a lo largo del tiempo, y a la ubicación epicárdica de algunos circuitos de reentrada no accesibles al mapeo endocárdico convencional y a la ablación con catéter34. Su indicación se reserva para pacientes con MCAVD que presentan un síncope con alta sospecha de ser de origen arrítmico y que además presenten arritmias ventriculares (extrasístoles y/o salvas no sostenidas o sostenidas de origen ventricular) en exámenes como ECG, Holter o test de esfuerzo. También se utiliza en pacientes con TV refractarias a fármacos antiarrítmicos o en aquellos que tienen efecto colateral importante, o contraindicación para el uso de estos medicamentos. Vale la pena mencionar que esta terapia no es una alternativa al DAI. En este sentido, incluso se plantea su uso en los casos de MCAVD con DAI que tienen descargas apropiadas por TV, con el objetivo de evitar descargas del DAI, disminuir los fármacos y mejorar la calidad de vida del paciente.
Conclusiones
La MCAVD es una entidad cuya definición diagnóstica se encuentra en desarrollo en pediatría. El rol del especialista pediátrico que se enfrenta a la sospecha de esta patología es distinto al del médico que trata al enfermo joven y al adulto. En etapas precoces se deben distinguir anomalías eléctricas en el ECG o Holter y más adelante anomalías estructurales que comprometan el movimiento de la pared y aneurismas focales que afectan predominantemente al VD, en particular al área subtricuspídea. La presencia de una mutación causal se constituye actualmente como criterio mayor, por lo que habilita al diagnóstico definitivo en presencia de algunas características adicionales. Esto ha aumentado la sensibilidad diagnóstica y ha reducido la prevalencia de menores de 18 años considerados no afectados de un 62% a un 53%. Por lo tanto, el inicio de la valoración del paciente pediátrico con un familiar enfermo portador de un gen mutado debe ser la evaluación genética. La utilización de pruebas genéticas en cascada ha demostrado mejorar la protección que podemos ofrecerles a los pacientes a largo plazo.

