











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
Los avances en las últimas décadas han aportado mucha información sobre la etiología y la historia natural de la miocardiopatía dilatada (MCD). Lo que hasta hace algunos años era catalogado genéricamente como MCD idiopática, hoy tiene tres o cuatro nombres distintos, basados fundamentalmente en los avances en las imágenes de la mano de la resonancia magnética y de la tipificación que ofrece la genética. Esta mejora en la definición de las etiologías ha permitido entender que las evoluciones pueden ser distintas, así como los tratamientos, los pronósticos y la necesidad de estudios familiares. En relación a este último concepto pasamos de evaluar pacientes a evaluar familias.
Revisión del tema
Definición
Constituye una causa importante de morbimortalidad cardiovascular por insuficiencia cardíaca congestiva o arritmias. Estudios de prevalencia estiman por lo menos tasas de 2%-3% de disfunción sistólica del ventrículo izquierdo (VI) y de 1,5% de insuficiencia cardíaca congestiva (ICC) en la población general. Su prevalencia en adultos se estima en alrededor de 1/2.500 individuos.
La definición requiere la presencia de dilatación ventricular izquierda con disfunción sistólica en ausencia de condiciones de carga que puedan justificarlo (hipertensión, enfermedad valvular o enfermedad coronaria). Es importante resaltar que estas condiciones pueden estar presentes, pero no ser suficientes para justificar el grado de disfunción o dilatación.
La disfunción sistólica se define por la fracción de eyección del VI anormal, medida usando cualquier modalidad y evidenciada por dos técnicas de imagen independientes o en dos ocasiones distintas por la misma técnica, de preferencia ecocardiograma o resonancia magnética.
La dilatación del VI se define por el aumento de los volúmenes telediastólicos, con valores que superen los dos desvíos estándar del valor normal según nomograma (puntajes corregidos por superficie corporal, edad y sexo)1,2.
Etiologías
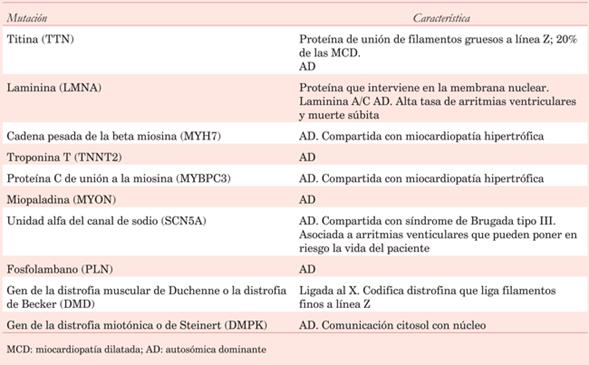
La Sociedad Europea de Cardiología clasifica las MCD dividiéndolas en genéticas o no genéticas, entendiendo que además existe una interacción entre los factores extrínsecos o ambientales que modulan la genética. El hallazgo en estudios poblacionales de una alta tasa de familiares afectados (20% a 50%) de pacientes portadores de MCD, hace sospechar un importante componente de herencia genética. En la mayoría de casos las mutaciones presentan una herencia autosómica dominante, aunque también las hay con formas recesivas o ligadas al cromosoma X. La mayoría se encuentran en los genes que codifican estructuras del sarcómero, seguidos por las estructuras de los puntos de unión celular y de la membrana nuclear. En algunos casos se ha detectado la presencia de más de una mutación, que pueden no estar en todos los individuos del grupo familiar. Esto puede proporcionar una explicación para la variación en la penetrancia individual vista en algunas familias. En la (tabla 1) se resumen las mutaciones más frecuentemente halladas2,3.
Causas no genéticas
Tóxicas
Varios compuestos químicos pueden inducir MCD; los más comunes son el consumo excesivo de alcohol y quimioterápicos. La miocardiopatía alcohólica es a menudo subdiagnosticada, ya que algunos estudios muestran un prevalencia de hasta el 20% de las MCD. El alcohol causa la disfunción sistólica del VI de una manera aparentemente relacionada con la dosis y algunos estudios sugieren reversibilidad tras la abstención4. En el caso de las drogas de quimioterapia existen tres tipos diferentes. Por un lado, la cardiotoxicidad tipo I (antraciclinas) y por tirosín-quinasa (imatinib); en estas el mecanismo pasa por necrosis celular, siendo por ende irreversible y asociada a mal pronóstico. Por otro lado, se encuentra la cardiotoxicidad tipo II (trastuzumab); la misma afecta la función de los miocitos sin provocar muerte celular, por lo que tiende a ser reversible y de mejor pronóstico. En algunos casos los efectos son muy cercanos a la aplicación y en otros lleva más tiempo desarrollarlos. Es importante destacar que los pacientes que desarrollan MCD provocada por una terapia oncológica suelen presentar hasta 3,5 veces más mortalidad que los pacientes con diagnóstico de MCD idiopática. Tanto en el alcohol como en algunas drogas quimioterápicas parece existir una susceptibilidad individual que se relaciona con mecanismos genéticos y no genéticos.
Otros tóxicos descritos, como la cocaína y las anfetaminas, pueden dañar el miocardio a través del vasoespasmo5.
Metabólicas
Las hormonas tiroideas ejercen efectos sobre la contracción y la relajación del corazón y los estudios experimentales sugieren fuertemente que intervienen en vías intracelulares fundamentales para la preservación de la estructura cardíaca y del rendimiento funcional del corazón, tanto en condiciones normales como patológicas. Influyen en la función diastólica y sistólica; además, la función contráctil del ventrículo se modifica según los cambios hemodinámicos asociados con los efectos de estas hormonas sobre el tono vascular periférico. Los trastornos tiroideos se asocian con aparición y progresión de la insuficiencia cardíaca. El hipotiroidismo y el hipertiroidismo pueden ocasionar daño cardíaco. Los estudios bioquímicos hormonales son importantes para diferenciar la insuficiencia cardíaca del hipotiroidismo, ya que las manifestaciones clínicas (disnea, edemas y derrame pleural) y los hallazgos electrocardiográficos (cambios en la onda T), y ecocardiográficos (menor contractilidad y dilatación cardíacas) pueden ser similares. El hipotiroidismo puede asociarse con hipercolesterolemia y aterosclerosis e induce fibrosis cardíaca mediante la estimulación de los fibroblastos; en el hipertiroidismo se observan los cambios opuestos. Tanto en casos de hipo como de hipertiroidismo el tratamiento mejora el cuadro, pudiendo llegar a la normalidad.
El concepto de MCD diabética específica continúa siendo objeto de debate, dado que la insuficiencia cardíaca en los pacientes diabéticos se asocia con frecuencia a hipertensión arterial, enfermedad coronaria, falla renal, siendo difícil desligar el daño miocárdico ocasionado por estas tres patologías del producido por la diabetes en sí misma. Son conocidos los fenómenos de falla diastólica, fibrosis intersticial, hipertrofia miocítica, disfunción microvascular y autonómica, ligados a la diabetes. El control metabólico estricto colabora con la reducción del impacto miocárdico de la enfermedad.
La hemocromatosis por acumulación de hierro y la enfermedad de Wilson por acumulación de cobre son poco frecuentes5,6.
Los trastornos del eje suprarrenal pueden inducir alteraciones miocárdicas, fundamentalmente los cuadros de feocromocitoma, Addison o Cushing. Otras alteraciones que deben ser descartadas son la hipocalcemia crónica y la hipokalemia.
Inflamatorias
La miocarditis se define como una enfermedad inflamatoria del músculo cardíaco y es una causa importante de insuficiencia cardíaca aguda y muerte súbita. Es hoy por hoy una de las causas más frecuentes de MCD. El diagnóstico históricamente se ha basado en la sospecha médica y la presentación clínica, combinada con datos de laboratorio y parámetros presentes en la biopsia endomiocárdica (BEM) que confirma dicho diagnóstico (por histología, inmunohistología y evidencia molecular). La BEM proporciona un diagnóstico etiológico definitivo que puede conducir a tratamientos específicos, como los antivirales o los inmunosupresores. En los últimos años, los avances de técnicas no invasivas, como la resonancia magnética cardíaca, han sido de gran utilidad para respaldar el diagnóstico de miocarditis y aunque se ha establecido que el diagnóstico definitivo es por BEM, no se realiza con frecuencia por razones de seguridad7-9.
Los virus son la causa de la mayoría de los casos de miocarditis o miocardiopatía inflamatoria y pueden inducir una respuesta inmunitaria causante de inflamación pese a haberse eliminado el patógeno. El sida es reconocido como una causa importante de miocardiopatía dilatada, que se ha logrado reducir sustancialmente con la introducción de la terapia antirretroviral hiperactiva (TRH). La mortalidad en estos casos es muy superior a la de otras miocardiopatías. El virus parece infectar los miocitos en distribución de islotes. Sin embargo, se desconoce cómo el HIV puede entrar a las células receptoras negativas de CD4, como son los miocitos. También se han descrito miocarditis por hepatitis C y B. La miocardiopatía chagásica es una forma de miocarditis en sus inicios, representando una causa muy frecuente en Latinoamérica, a la que pocas veces diagnosticamos en fase aguda10.
La autoinmunidad cardíaca participa en la patogénesis de la miocardiopatía por HIV, ya que hasta en 30% de los pacientes se encuentran autoanticuerpos cardioespecíficos. Varios estudios han demostrado que los pacientes con encefalopatía son más propensos a morir de insuficiencia cardíaca que los que no presentan encefalopatía. Las células del miocardio y del cerebro que no son susceptibles a ningún tratamiento, pueden contener el HIV en sus superficies y liberar citoquinas tóxicas. Otro factor que induce el desarrollo de insuficiencia cardíaca en estos pacientes es el déficit nutricional, particularmente de oligoelementos.
La miocarditis puede ser reversible si el proceso inflamatorio agudo sana y la causa se resuelve, pero en hasta 30% de los casos puede progresar a MCD, por lo que es muy importante seguir a los pacientes, más allá de que inicialmente los síntomas retrograden.
Miocardiopatía periparto
La miocardiopatía periparto es poco frecuente, pero puede poner en riesgo la vida de la paciente. Es un trastorno definido por el desarrollo inexplicable de insuficiencia cardíaca sistólica hacia el final del embarazo o en el puerperio. La relevancia de esta entidad radica en la alta morbimortalidad que genera tanto para la madre como para el hijo, sobre todo en aquellos casos donde se realiza diagnóstico en forma tardía y en consecuencia se demora el inicio del tratamiento adecuado.
Suelen existir ciertas características de la paciente que se asocian a mayor riesgo de padecer dicha patología: edad avanzada, multiparidad e hipertensión con o sin preeclampsia. La etiología es compleja e incluye autoinmunidad, microquimerismo fetal, infección por virus, citoquinas activadas por estrés y toxicidad causada por un producto de escisión anormal de prolactina. Como en otras causas aparentemente adquiridas de MCD, la predisposición genética parece importante en algunos casos11.
En cuanto a la recuperación de la función ventricular, las pacientes con diagnóstico de miocardiopatía periparto presentan una mayor tasa de recuperación que las pacientes con otras formas de miocardiopatía dilatada, ya que se ha visto que la normalización de la función del VI a los seis meses varía entre 23% y 54% de los casos, dependiendo de las series publicadas, existiendo una menor tasa de recuperación tanto en las mujeres afroamericanas, según estudios realizados en Estados Unidos, como en mujeres africanas, en comparación con el mayor porcentaje que se evidencia en las mujeres de raza blanca.
Actualmente se establece que las mujeres que vuelven a quedar embarazadas tienen peor pronóstico que las que no presentaron la enfermedad, con una mayor incidencia de insuficiencia cardíaca en los siguientes embarazos y una mayor mortalidad en los casos en que la función ventricular sistólica no se ha recuperado totalmente, y aun con una recuperación total también persiste cierto riesgo.
Herramientas para el diagnóstico
Entendemos que es importante tener una estrategia diagnóstica cuando nos enfrentamos por primera vez con un paciente presuntamente afectado. Una cuidadosa historia clínica con examen físico, electrocardiograma y radiografía de tórax, continúan siendo fundamentales, más allá de disponder de estudios de mayor complejidad.
Anamnesis
El interrogatorio debe ser minucioso. Debe incluir los factores de riesgo cardiovascular, especialmente hipertensión arterial, antecedentes familiares de enfermedad cardíaca y muerte súbita y antecedentes cardiovasculares del paciente: enfermedad coronaria con revascularización, enfermedad valvular, arritmias asociadas. Por otro lado, siempre se debe interrogar sobre la exposición a fármacos cardiotóxicos, quimioterápicos y radiación. Se debe realizar especial hincapié en lo que respecta a los diferentes síntomas del paciente, detallando la secuencia de presentación de los mismos, la asociación con fiebre, con dolores torácicos, articulares, etcétera12,13).
Radiografía de tórax
Hoy día la radiografía de tórax tiene poco uso en el proceso diagnóstico de los pacientes con MCD. Suele solicitarse en el estudio inicial para descartar patologías respiratorias asociadas y para el seguimiento si el paciente comienza con síntomas de insuficiencia cardíaca. Sin embargo, la radiografía torácica suele mostrar un índice cardiotorácico aumentado y podría mostrar congestión o edema venoso pulmonar. En muchos casos suele encontrarse derrame pleural bilateral o asimétrico.
Laboratorio
En la evaluación inicial se recomienda la realización de un laboratorio completo que incluya hemograma con leucocitos y plaquetas, ionograma, función renal, hepatograma y coagulograma. En las guías actuales se recomienda la solicitud del péptido natriurético tipo B (BNP o pro BNP NT), ya que puede ser utilizado como marcador de pronóstico una vez que el paciente desarrolla insuficiencia cardíaca. La anemia es frecuente en la MCD, presente en hasta el 25% de los pacientes y constituye un factor de mal pronóstico. Es necesaria la realización de serología para Chagas y para virus HIV, hepatitis C y B. Estudios específicos, como dosaje de ferritina, cortisol o catecolaminas, pueden orientarse según sospecha.
Electrocardiograma
El electrocardiograma no presenta patrones específicos en la MCD, aunque no suele ser normal. Existen ciertos criterios electrocardiográficos que pueden guiar cuando se tiene una alta sospecha de la patología. Uno de ellos es que el 30% de los pacientes que tienen MCD presentan un bloqueo completo de rama izquierda. El hallazgo de fibrilación auricular u otras arritmias suele asociarse con mal pronóstico. La presencia de ondas Q o resaltos, o mala progresión de Rs suele asociarse con patología coronaria preexistente.
Ecocardiograma
La ecocardiografía transtorácica (ETT) es la técnica de elección para evaluar la función miocárdica sistólica y diastólica de los ventrículos derecho e izquierdo. La identificación de anomalías en la motilidad regional de la pared puede ser particularmente importante en pacientes con alta sospecha de enfermedad coronaria o miocarditis.
La ETT debería evaluar la dilatación del VI con medición de diámetros de fin de diástole y fin de sístole, así como volúmenes ventriculares, masa del VI y espesores miocárdicos, describiendo con la mayor exactitud posible la distribución de las zonas de aumento o adelgazamiento del espesor miocárdico. La fracción de eyección debe ser medida tomando la mayor cantidad de vistas posibles, ya que es el parámetro, junto con los volúmenes ventriculares, que usaremos para seguimiento. Siempre debe llevarse a cabo una adecuada evaluación de la presencia de valvulopatías tratando de definir si son secundarias a la dilatación ventricular o si son primarias y probable causa de la miocardiopatía dilatada. La medición de parámetros de función diastólica como llenado ventricular, tamaño auricular, relación E/e’ como expresión de presiones de llenado y presión sistólica pulmonar no debe ser olvidada. La mayor disponibilidad en los equipos de tecnología de Doppler tisular así como speckle tracking facilita la caracterización tisular no invasiva2,4.
La ecocardiografía tridimensional mejora la cuantificación de los volúmenes de las cavidades y el cálculo de la función sistólica del VI. La gran limitación es que requiere de un médico especializado, capacitado y con experiencia en la práctica, y que depende mucho de la calidad de la ventana ultrasónica del paciente.
No se recomienda la utilización de la ecocardiografía transesofágica de rutina; sin embargo, puede ser útil en algunos contextos clínicos para pacientes con enfermedad valvular o cardiopatías congénitas.
Tomografía computada
La angiotomografía coronaria computada permite la evaluación no invasiva de la anatomía coronaria en pacientes con una probabilidad pretest baja de enfermedad coronaria, evitando de esta forma la realización de una cinecoronariografía y salvando las limitaciones de los estudios como ergometría, ecoestrés o SPECT en pacientes con electrocardiograma patológico y enfermedad miocárdica. En algunas ocasiones, en que los pacientes no toleren o no puedan realizarse una resonancia cardíaca, la tomografía puede colaborar para la caracterización de los espesores parietales.
Estudio de hemodinamia
Se utiliza para descartar que la etiología de la MCD sea la enfermedad coronaria, cuando no pudo efectuarse por otro método no invasivo. La realización de cateterismo derecho con medición de presiones y gasto, colabora en la comprensión de la hemodinamia y es necesaria en pacientes en etapa avanzada para evaluación de trasplante cardíaco.
Resonancia cardíaca
La resonancia magnética cardíaca (RMC) es una técnica de imagen de gran utilidad en la evaluación de pacientes con miocardiopatías. El aspecto más relevante de la RMC es la caracterización tisular para la identificación de la fibrosis mediante las imágenes de realce tardío con gadolinio (RTG). La determinación de la fibrosis extendió las aplicaciones de la RMC para evaluar pacientes con infarto de miocardio, determinar la viabilidad miocárdica y evaluar miocardiopatías de origen no isquémico14. El RTG es un elemento esencial en el proceso patogénico de varias miocardiopatías. A diferencia de otros tejidos, el miocardio tiene una limitada capacidad de regeneración ante una noxa, mientras que el proceso de reparación se lleva a cabo mediante el desarrollo de fibrosis. Es por ello que está asociada con un peor pronóstico, por mayor remodelado ventricular, desarrollo de insuficiencia cardíaca, hospitalización y muerte súbita. Existen dos tipos de fibrosis, la intersticial que consiste en una fibrosis difusa que no se puede identificar en las secuencias de realce tardío y una fibrosis de reemplazo focal donde los miocitos son reemplazados por tejido fibroso.
La disposición de este RTG permite la identificación de distintas etiologías de miocardiopatías. Los patrones de realce tardío más comúnmente hallados son: sin realce (59%), realce lineal mesocárdico (28%) y realce subendocárdico o transmural (13%). El ejemplo característico es el de las miocarditis, donde se presenta RTG a nivel subepicárdico o intramiocárdico sin corresponder a un territorio coronario, en contraposición con los casos de origen coronario que son subendocárdicos o transmurales y se corresponden con un territorio vascular.
Para realizar una correcta evaluación hay que tener en cuenta que el 60%-70% de los pacientes con MCD idiopática no presenta RTG, ya que las mutaciones no generan alteraciones intersticiales. A diferencia de la miocardiopatía hipertrófica donde la fibrosis está siempre presente, en la MCD idiopática está presente en etapas más avanzadas de la enfermedad.
Por otro lado, la presencia de realce tardío puede ayudar a identificar pacientes que se beneficiarán con la colocación de un cardiodesfibrilador implantable en prevención primaria, sobre todo aquellos con fracción de eyección menor a 35%, con miocardiopatía no isquémica. Es importante tener en cuenta las zonas con fibrosis, ya que pueden tener un rol importante en aquellos pacientes en los que se evalúa la terapia de resincronización15.
Otra ventaja de la RMC es la correcta evaluación del ventrículo derecho, que resulta difícil en la ecocardiografía debido a su forma compleja, que no se corresponde con ningún modelo geométrico.
Estudio genético
La MCD es una causa común de insuficiencia cardíaca. Cuando la etiología de la miocardiopatía no se logra determinar, se llama MCD idiopática; en este grupo es en el que la etiología genética tiende a tener más importancia.
Hoy día se sabe que entre el 20%-35% de las MCD tienen una asociación familiar. Por esta razón, el interrogatorio de los pacientes es tan importante y debe ser minucioso. Se debe incluir en el mismo al menos cuatro generaciones previas. Una vez identificada la asociación, se recomienda un screening clínico a los diferentes miembros de la familia. La mayoría de las MCD son transmitidas con un patrón autosómico dominante existiendo otros patrones menos frecuentes. No se recomienda realizar una evaluación genética si no aparece ningún tipo de asociación familiar.
A diferencia de la miocardiopatía hipertrófica donde se encuentran principalmente afectados los sarcómeros, la MCD es más heterogénea, afectando proteínas con diferentes roles dentro de la célula. Actualmente, más de 50 genes se encuentran asociados a la MCD. Existen múltiples clasificaciones. Por un lado se pueden clasificar por las mutaciones en las funciones celulares: sarcoméricas: TTN, MYH7, ACTC1, TPM1 y TNN13; aquellas que afectan el disco-Z: BAG3. Mutaciones que afectan la homeostasis electrolítica: PLN (phospholamban), SERCA2a, SCN5A, R222Q. Mutaciones que alteran la estructura proteica: LMNA. EMD, SYNE1, EDMD1, DES. Por último, mutaciones que afectan la expresión génica: RBM20, EYA48.
Por otro lado, se pueden clasificar en asociados a enfermedad de la conducción cardíaca y no asociados. Dentro del primer grupo vamos a encontrar al gen de la LMNA y al gen SCN5A. Y los genes no asociados a enfermedades de la conducción son: TNN, MYH7, TNNT2, LAMA4, VCL, ABCC9, etcétera. La afección más frecuente son las mutaciones del gen de titina (TNN), que es una proteína estructural de las miofibrillas. En general, las mutaciones afectan a las proteínas de las miofibrillas intracelulares sin comprometer el intersticio5.
Existen otras miocardiopatías que también tienen una asociación genética. Se ha visto que la cardiomiopatía posparto se encuentra muy ligada al gen TNN. También se encuentran miocardiopatías dilatadas mediadas inmunológicamente u asociadas a distrofias musculares, que son menos frecuentes.
Uno de los roles fundamentales del test genético en la MCD es el screening de familiares de primer grado de pacientes a los que se les ha diagnosticado la enfermedad; de esta forma se logran detectar etapas tempranas de la enfermedad y por ende se mejoran las tasas de sobrevida. Se considera una MCD familiar cuando hay al menos dos familiares con diagnóstico de la enfermedad o un familiar de primer grado presenta muerte súbita siendo menor de 35 años. La primera aproximación a los familiares directos debe ser un electrocardiograma y un ecocardiograma. Pero posteriormente deben tener un adecuado seguimiento, ya que es muy variable la expresión de la enfermedad y la penetrancia del gen enfermo. La edad de aparición de la patología suele ser entre los 20 y 50 años y raramente se diagnostica en ancianos, pero la severidad fenotípica y la edad de aparición suelen variar en los diferentes integrantes de la familia. Inclusive se ha visto que no todos los miembros de la familia que uno esperaría presentan dicha mutación, ya que en ciertos casos suele mostrar “reducida” penetrancia. Identificar los factores ambientales o genes modificadores que afectan la expresión fenotípica nos permitirá entender el mecanismo detrás de la MCD familiar2).
El consejo genético incluye asesoramiento reproductivo, profesional y deportivo. El equipo asesor debe contar con profesionales experimentados. Se debe explicar con claridad a pacientes y familiares los riesgos y beneficios del test, así como las implicaciones clínicas, psicológicas y sociales. Idealmente, el consejo reproductivo debe realizarse antes de la gestación. Se deben tratar aspectos como patrón de herencia, penetrancia, expresividad e historia familiar. Se debe informar de las alternativas reproductivas a todo sujeto portador de una mutación causal.
Conclusiones
La evolución del estudio de las MCD ha llevado a que el rótulo de idiopática quede cada vez más relegado. La genética y las imágenes de alta resolución serán las responsables del avance en los próximos diez años y deberemos usarlas cuando sea necesario. Hoy debemos aplicar un dogma que aprendimos con la miocardiopatía hipertrófica: “Al enfrentarnos con un paciente, lo estamos haciendo con una familia entera”, y debemos evaluar a pacientes asintomáticos. Este cambio de mentalidad mejorará significativamente el impacto de nuestra práctica clínica. A la pregunta del título sobre cuándo debemos evaluar etiología, la respuesta es contundente: siempre.

