











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkLa muerte súbita cardíaca (MSC) en población infantil está causada principalmente por cardiopatías congénitas estructurales y enfermedades de origen genético como las miocardiopatías y las canalopatías1,2. En concreto, las canalopatías engloban un grupo de trastornos eléctricos primarios del corazón, sin alteraciones anatómicas apreciables, pero que predisponen a sufrir arritmias ventriculares polimórficas y MSC. Entre ellas, se incluyen el síndrome de QT largo, el síndrome de Brugada, la taquicardia ventricular polimórfica catecolaminérgica (TVCP) y el síndrome de QT corto. Los genes más frecuentemente asociados a estos trastornos codifican canales de sodio, potasio y calcio involucrados en diferentes fases del potencial de acción cardíaco. La aparición de técnicas de secuenciación masiva como next generation sequencing ha aumentado la rentabilidad del test genético para identificar mutaciones probablemente causales en las cardiopatías hereditarias3-5.
Recientemente la aplicación de estas técnicas en niños con canalopatías de etiología no aclarada ha permitido el descubrimiento de mutaciones en genes que codifican la calmodulina (CaM), una ubicua proteína de unión al calcio intracelular6-8. Casi todas las mutaciones identificadas impiden la unión de la proteína al calcio y la incapacita para modular dianas moleculares específicas en el corazón. Ello provoca una predisposición a arritmias ventriculares polimórficas por diferentes mecanismos, motivo por el que causa MSC a edades muy precoces. Es reseñable el grado de conservación tan elevado que presenta la proteína CaM, sin cambios en aminoácidos desde la aparición de los vertebrados. Relativamente pequeña, está formada por un dominio globular N-terminal y otro C-terminal, casi simétricos, separados por una hélice alfa. Cada uno de los dominios contiene dos lugares de unión al calcio formados por 12 residuos rodeados por dos hélices9.
La fisiopatología mediante la cual mutaciones en calmodulina producen alteración de la función de los canales iónicos y fenotipo de canalopatía cardíaca no es bien conocida. Existen datos que sugieren una mala regulación de la apertura y cierre de los canales de sodio y potasio de la membrana celular, respectivamente, conllevando una prolongación por este motivo del potencial de acción y del intervalo QT del electrocardiograma (ECG). Sin embargo, datos recientes sugieren que la corriente afectada es de calcio, motivado por la falta de inactivación de canales de Cav 1.2 tipo L durante la fase 2 del potencial de acción, condicionando una entrada excesiva de calcio en esta fase y prolongando el potencial de acción10-14. Otras mutaciones, quizá las más alejadas del cuarto dominio y del extremo C-terminal, como la N98S y la N53I, producen un fenotipo más consistente con TVCP15, con extrasistolia pleomórfica con el ejercicio y taquicardia ventricular bidireccional. Algunos autores han estudiado el mecanismo molecular que conlleva la aparición de pospotenciales, siendo la hipótesis más plausible la activación excesiva del canal ryanodina por la CaM unida de forma defectuosa al calcio16, y motivando por este motivo una sobrecarga diastólica de calcio citoplasmático, que es la anomalía eléctrica de base de los pospotenciales tardíos observados en la TVCP. En resumen, la CaM regula procesos intracelulares esenciales para la correcta generación del potencial de acción cardíaco y la contracción del miocardiocito, por lo que los mecanismos molecu lares para la provocación de arritmias son diversos (Figura 1).
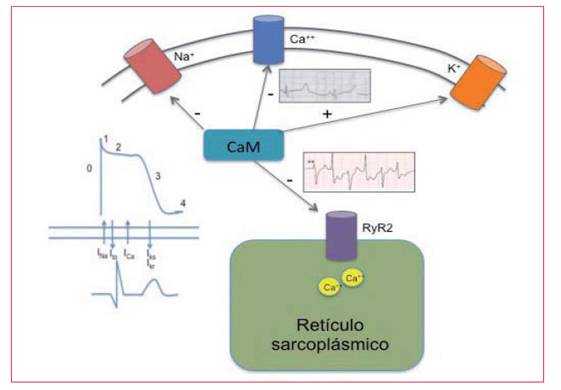
Figura 1: Esquema representativo de la acción moduladora de la calmodulina sobre canales de la membrana plasmática y del retículo sarcoplásmico. La calmodulina disminuye la disponibilidad de canales de sodio y favorece la inactivación de canales de calcio, mientras que favorece las corrientes repolarizadoras de potasio. A nivel de la ryanodina promueve el cierre de estos canales para evitar la sobrecarga citosólica de calcio.
Existen tres genes que codifican dicha proteína situados en diferentes cromosomas: CALM1, CALM2, y CALM3, con secuencias de nucleótidos que completan los 149 aminoácidos que la componen, idénticas entre sí. Las tres proteínas se expresan en el corazón, aunque quizá con mayor intensidad CALM36. Interesantemente, CALM3 es el gen que con menor frecuencia se ha asociado a enfermedades eléctricas primarias y arritmias, y los escasos casos descritos son especialmente severos17. Ello hace pensar que mutaciones en este gen son poco compatibles con la vida. Sin embargo, ha sido en CALM1 y CALM2 donde se ha descrito la mayoría de mutaciones, todas ellas con un escenario clínico similar: edad muy temprana de presentación (habitualmente antes de los 5 años de edad), prolongación acusada del intervalo QTc en mutaciones más cercanas al extremo C-terminal de la proteína, y fenotipos compatibles con TVCP en aquellas mutaciones del dominio N-terminal, frecuente presencia de bloqueo auriculoventricular 2:1 y alternancia de la onda T, y una alta incidencia de arritmias ventriculares malignas a edad muy precoz, incluso con muerte súbita. Este fenotipo común ha motivado que se clasifique la enfermedad desde el punto de vista molecular como calmodulinopatía, diferen ciándolo del resto de enfermedades eléctricas pri marias en la infancia por su etiología genética y su curso clínico particular.
Existen escasos datos de series de pacientes en la literatura, ya que la mayoría de artículos publicado han correspondido a casos clínicos aislados apoyados en elegantes estudios funcionales. Recientemente se ha presentado un trabajo en el último congreso de la American Heart Association donde se describen las consecuencias clínicas de las mutaciones en CALM en la serie más amplia hasta la fecha, denominada International Calmodulinopathy Registry (ICaMR)18. De los 49 pacientes portadores de mutaciones patogénicas en heterocigosis en cualquiera de los genes CALM, el 84% había presentado eventos arrítmicos al momento del estudio con una edad media de los mismos de cuatro años. Destaca la elevada incidencia de parada cardíaca o MSC, con 61% de los casos, muy superior a cualquier otro trastorno eléctrico primario en la infancia. Los fenotipos descritos fueron fundamentalmente síndrome de QT largo y taquicardia ventricular catecolaminérgica.
En resumen, las mutaciones en CALM provocan un fenotipo de canalopatía en niños con una especial gravedad y precocidad respecto a otras etiologías genéticas de enfermedades eléctricas primarias. Los genes CALM1-3 debes ser secuenciados en niños con canalopatías y estudio genético convencional negativo, y las medidas terapéuticas en portadores deben instaurarse de forma precoz para evitar eventos arrítmicos graves.

