










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Uruguaya de Cardiología
Print version ISSN 0797-0048On-line version ISSN 1688-0420
Rev.Urug.Cardiol. vol.31 no.1 Montevideo Apr. 2016
Caso clínico
Estratificación del riesgo de muerte súbita en un caso de miocardiopatía hipertrófica con fragmentación del complejo QRS
Dres. Oscar Pedemonte, Federico Ferrando-Castagnetto, Alicia Torterolo, Roberto Ricca-Mallada, Lic. NC Pablo Marichal
Cátedra de Cardiología. Centro Cardiovascular Universitario. Universidad de la República.
Correspondencia: Dr. Óscar Pedemonte, J. F. Zubillaga 1123 bis, Apto. 601, Montevideo, Uruguay.
Correo electrónico: oscar984_2@hotmail.com
Recibido Dic 9, 2015; aceptado Mar 12, 2016.
Palabras clave:
INFORMES DE CASO
ESTRATIFICACIÓN DEL RIESGO
MUERTE SÚBITA
CARDIOMIOPATÍA HIPERTRÓFICA
Key words:
CASE REPORTS
RISK STRATIFICATION
DEATH SUDDEN
CARDIOMYOPATHY, HYPERTROPHIC
Introducción
La miocardiopatía hipertrófica (MCH) es una entidad de base genética caracterizada por un aumento del grosor parietal miocárdico sin causa aparente(1). Constituye una cardiopatía congénita muy frecuente y la principal causa de muerte súbita (MS) en atletas menores de 35 años(2), con una prevalencia estimada en 0,2%. Se presenta en un espectro anátomo-clínico y pronóstico muy variable, que va desde un grosor parietal del ventrículo izquierdo (VI) casi normal hasta una hipertrofia masiva, y desde casos con pronóstico similar a la población general hasta individuos jóvenes asintomáticos que debutan con un desenlace fatal. Se diagnostica mediante la detección de un aumento en el grosor del VI ³15 mm en uno o más segmentos que no puede explicarse únicamente por condiciones de sobrecarga; aunque en los familiares de primer grado con MCH inequívoca el grosor parietal requerido para confirmar el diagnóstico es aún menor(1). Si bien mucho se ha avanzado en la caracterización precisa de su genética, anatomía y fisopatología, la detección oportuna y exacta de aquellos pacientes con MCH en mayor riesgo de evolución desfavorable sigue siendo un desafío clínico.
Describimos el caso de un hombre joven asistido en la policlínica de MCH del Centro Cardiovascular Universitario que exhibía importante fragmentación del QRS en el electrocardiograma (ECG), individualizando precozmente la estratificación de riesgo de MS con los aportes de un score de riesgo de reciente validación.
Caso clínico
Paciente de sexo masculino, 21 años, sedentario, fumador intenso, exconsumidor de cocaína. No relata cifras elevadas de presión arterial (PA). Como antecedentes familiares destacaba la noción de padre, tío y hermano portadores de MCH, motivo por el cual concurre a control cardiológico ambulatorio. No presentaba antecedentes de MS en la familia. Mientras deambulaba presentó un episodio de pérdida brusca y transitoria de conocimiento, de escasos minutos, con recuperación ad integrum, precedido de dolor precordial. Hasta entonces el paciente manifestaba episodios ocasionales de palpitaciones rápidas, breves, en reposo. Examen clínico en la emergencia: paciente lúcido, eupneico, buen estado nutricional, con hematoma palpebral izquierdo. Al examen cardiovascular central no se palpaba el latido apexiano, ritmo regular de 60 cpm, S1 y S2 de tonalidad normal, soplo en mesocardio de intensidad II/VI (Levine) sin irradiaciones, que no se modificaba con la maniobra de Vasalva. Al examen vascular periférico, PA: 125/80 mmHg, pulsos simétricos normales, ausencia de ingurgitación yugular. Los campos pulmonares estaban libres y el examen neurológico era normal. El ECG basal de 12 derivaciones mostraba ritmo sinusal y marcadas alteraciones en la depolarización y repolarización ventricular (figura 1). La curva de biomarcadores de lesión miocárdica (TnI) fue negativa y el resto de la analítica de laboratorio fue normal.
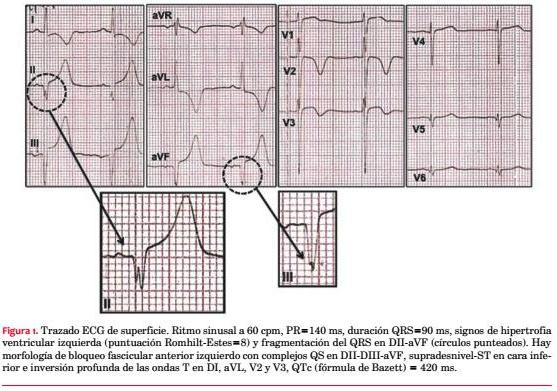
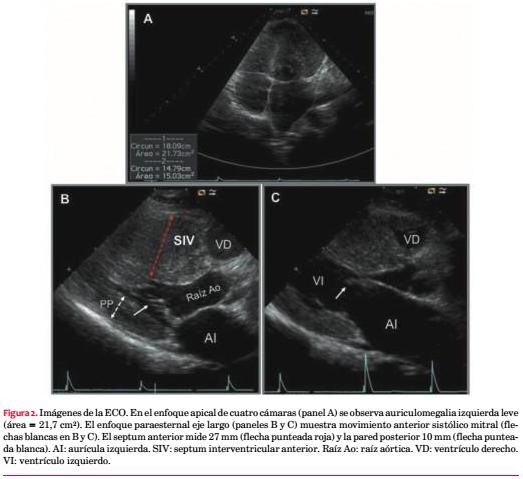
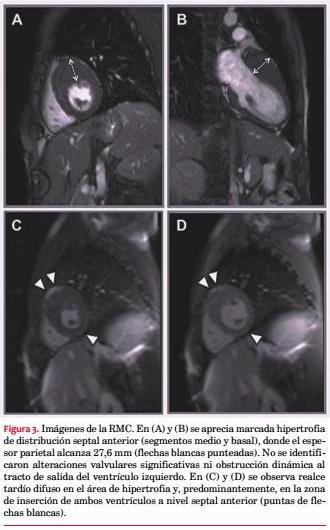
Comentarios
Describimos el caso de un hombre joven con familiares portadores de MCH que consulta por síncope precedido de dolor torácico, situación clínica que sugiere la etiología cardiovascular del mismo. Sabiendo que en el 60% de los casos la MCH responde a una causa genética de transmisión autosómica dominante, más comúnmente a una o varias mutaciones en los genes que codifican proteínas sarcoméricas, planteamos que el paciente sea portador de una MCH. Los pacientes con MCH que presentan obstrucción al tracto de salida del VI suelen exhibir un soplo eyectivo en toda el área precordial que suele aumentar con la disminución en la precarga al VI inducida por las maniobras de Valsalva y cuclillas-bipedestación(2). En nuestro caso no se confirmó el comportamiento dinámico del soplo.
El registro Holter y la pruega ergométrica graduada son técnicas accesibles y de bajo coste que cumplen un rol importante en la estratificación de riesgo(1,2); en el caso descrito no detectaron marcadores de riesgo elevado.
El enfoque terapéutico de la MCH se focaliza en dos grandes objetivos: tratar los síntomas y prevenir la MS. Este último punto obliga a estratificar precozmente el riesgo individual de MS en todos y cada uno los casos. Tradicionalmente, los factores de alto riesgo incluían el grosor parietal máximo >30 mm, la taquicardia ventricular no sostenida, el síncope de causa no aclarada, los antecedentes familiares de MS y la respuesta hipotensora al esfuerzo(1,2). La disponibilidad de algunos scores de riesgo representa un aporte novedoso y de potencial utilidad práctica en la estratificación temprana de la MCH. Estos scores son fácilmente accesibles al lado del paciente, están disponibles en dispositivos portátiles y tienen en cuenta varios factores de riesgo que son integrados como variables continuas y no dicotómicas, lo cual podría graduar mejor su peso individual. Dentro de estos scores destaca el MHC RISK-SCD , un modelo de predicción validado por O’Mahony y colaboradores en un gran estudio de cohortes retrospectivo(18). Esta herramienta permite estimar individualmente el riesgo de MS a cinco años basado en los marcadores tradicionales y en otras variables hasta ahora no consideradas como el gradiente de obstrucción al tracto de salida del VI, las dimensiones de la aurícula izquierda y la edad. El puntaje obtenido por el score estratifica el riesgo individual de MS en tres categorías: bajo (riesgo de MS a los cinco años <4%), moderado (4%-6%) y alto (> 6%). Su uso ha demostrado mejores beneficios en aquellos pacientes con uno o más factores de riesgo clásicos, disminuyendo la tasa de implantes de cardiodesfibrilador automático (DAI) innecesarios, pero mantiene un bajo poder predictivo en la subpoblación de pacientes sin ningún factor de riesgo clásico, en quienes se observa el 30% de las MS. Sin embargo, el MHC RISK-SCD SCORE aún no ha sido validado en pacientes menores de 16 años, deportistas de elite, pacientes sometidos a miectomía ventricular/ablación septal con alcohol o con enfermedades metabólicas infiltrativas. Con base en sus resultados, la más reciente recomendación de la ESC define una indicación de implante de DAI, Clase IIa, nivel de evidencia B, en los pacientes de riesgo elevado(1). En este caso, el MHC RISK-SCD al diagnóstico predijo un riesgo moderado de MS a los cinco años (5,89%), aunque muy cercano al riesgo elevado (figura 4). Razonando con base en los marcadores pronósticos tradicionales(1,2), el riesgo de MS está marcado principalmente por la presencia de síncope de causa no aclarada en un sujeto joven. En este contexto clínico, las guías de la Asociación Americana de Cardiología (2011) señalan que el implante de DAI representaría una alternativa “razonable”(19). En este caso se procedió al implante tras adicionar el riesgo asociado a la edad temprana y la fragmentación del QRS al peso pronóstico moderado (cercano a elevado) sugerido por el score MHC RISK- SCD.Tanto la detección de factores de riesgo adicionales como los aportes pronósticos diferenciales de los nuevos scores versus la consideración exclusiva de los marcadores más tradicionales merecen ser evaluados mediante estudios prospectivos.

Conclusiones
Reportamos el caso de un joven con MCH asimétrica anteroseptal que se presentó con síncope y marcadas alteraciones en el ECG, entre las que destaca la fragmentación del complejo QRS, un hallazgo asociado a la presencia de fibrosis miocárdica de potencial valor pronóstico. Estratificamos precozmente el riesgo de MS como moderado, con base en las recomendaciones internacionales y el MHC RISK-SCD SCORE. En este contexto clínico, decidimos proceder al implante de un DAI como medida de prevención primaria razonable. Aun disponiendo de modernas técnicas de imagen y nuevos scores de riesgo confiables al lado del paciente, el perfil evolutivo impredecible de la MCH a menudo dificulta la toma de decisiones terapéuticas.
Bibliografía
1. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35(39):2733-79.
2. Maron BJ. Miocardiopatia hipertrófica.En: Libby P, Bonow R Maan D et al. Braunwald Heart diseases, 8va edición, España, Elsevier, 2009;p.1763-73.
3. Harmon KG, Zigman M, Drezner JA. The effectiveness of screening history, physical exam, and ECG to detect potentially lethal cardiac disorders in athletes: a systematic review/meta-analysis. J Electrocardiol 2015;48(3):329-38.
4. Savage DD, Seides SF, Clark CE, Henry WL, Maron BJ, Robinson FC, et al. Electrocardiographic findings in patients with obstructive and nonobstructive hypertrophic cardiomyopathy. Circulation 1978;58:402-408.
5. Pelliccia A, Di Paolo FM, Corrado D, Buccolieri C, Quattrini FM, Pisicchio C, et al. Evidence for efficacy of the Italian national pre-participation screening programme for identification of hypertrophic cardiomyopathy in competitive athletes. Eur Heart J 2006;27(18):2196-200.
6. Perez Riera AR, Barbosa Barros R. Hypertrophic cardiomyopathy: value of electrocardiogram for the diagnosis of different types and for differential diagnosis with athlete’s heart. Rev Fed Arg Cardiol 2015;44(1):12-24.
7. Dumont CA, Monserrat L, Soler R, Rodríguez E, Fernandez X, Peteiro J, et al. Interpretation of electrocardiographic abnormalities in hypertrophic cardiomyopathy with cardiac magnetic resonance. Eur Heart J 2006;27(14):1725-31.
8. Song BG, Yang HS, Hwang HK, Kang GH, Park YH, Chun WJ, et al. Correlation of electrocardiographic changes and myocardial fibrosis in patients with hypertrophic cardiomyopathy detected by cardiac magnetic resonance imaging. Clin Cardiol 2013;36(1):31-5.
9. Grall S, Biere L, Clerfond G, Mateus V, Prunier F. ECG characteristics according to the presence of late gadolinium enhancement on cardiac MRI in hypertrophic cardiomyopathy Open Heart 2014; 5;1(1):e000101.
10. Montgomery JV, Harris KM, Casey SA, Zenovich AG, Maron BJ. Relation of electrocardiographic patterns to phenotypic expression and clinical outcome in hypertrophic cardiomyopathy. Am J Cardiol 2005;96(2):270-5.
11. Kawasaki T, Harimoto K, Honda S, Sato Y, Yamano M, Miki S, et al. Notched QRS for the assessment of myocardial fibrosis in hypertrophic cardiomyopathy. Circ J 2015;79(4):847-53.
12. Konno T, Hayashi K, Fujino N, Oka R, Nomura A, Nagata Y, et al. Electrocardiographic QRS fragmentation as a marker for myocardial fibrosis in hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol 2015;26(10):1081-7.
13. Nomura A, Konno T, Fujita T, Tanaka Y, Nagata Y, Tsuda T, et al. Fragmented QRS predicts heart failure progression in patients with hypertrophic cardiomyopathy. Circ J 2015;79(1):136-43.
14. To AC, Dhillon A, Desai MY. Cardiac Magnetic Resonance in hypertrophic cardiomyopathy. JACC Cardiovasc Imaging 2011;4(10):1123-37.
15. Maron MS, Maron BJ, Harrigan C, Buros J, Gibson CM, Olivotto I, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol 2009;54(3):220-8.
16. Green JJ, Berger JS, Kramer CM, Salerno M. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging 2012;5:370-377.
17. O’Hanlon R, Grasso A, Roughton M, Moon JC, Clark S, Wage R, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010;56: 867–874.
18. O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD); Hypertrophic Cardiomyopathy Outcomes Investigators. Eur Heart J 2014;35(30):2010-20.
19. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011;124:e783-e831.

