










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Uruguaya de Cardiología
Print version ISSN 0797-0048On-line version ISSN 1688-0420
Rev.Urug.Cardiol. vol.30 no.3 Montevideo Dec. 2015
Artículo original
Registro Uruguayo de Miocardiopatía Hipertrófica (RUMHI)
Dres. Inés Vidal, Jorge Estigarribia, Álvaro Báez, Luis Vidal
Cardiólogos. Coordinadores del Registro Uruguayo de Miocardiopatía Hipertrófica. Sociedad Uruguaya de Cardiología.
Correspondencia: Dra. Inés Vidal. Presidente Giró 2540. Montevideo CP 11600. Correo electrónico: invi@montevideo.com.uy
Recibido octubre 20, 2015; aceptado noviembre 15, 2015.
Resumen
Fundamento y objetivos: no contamos con datos clínicos, morfológicos, de manejo ni pronósticos de la miocardiopatía hipertrófica (MH) en Uruguay. Para obtenerlos, implementamos un Registro Uruguayo de MH (RUMHI).
Método: se estudió una población de 52 pacientes con MH, con seguimiento promedio de 31,7 ± 12,5 meses.
Resultados: con predominio femenino (58%) y edad de 6 a 82 años (50,3 ± 20,7), el 69% presenta síntomas, preponderando la disnea (54%), que se asoció a la presencia de obstrucción intraventricular (p=0,048) y el síncope a los antecedentes familiares de muerte súbita (p=0,033); 13% presenta fibrilación auricular. Veinticinco (48%) asocian hipertensión arterial (HTA); 23 de grado 1. Tienen patrón ecocardiográfico típico: cavidad pequeña, función sistólica conservada, hipertrofia ventricular izquierda (HVI) asimétrica (71%) y máximo espesor en los segmentos basales del septum (90%). El 58% presenta obstrucción dinámica; se pesquisó la latente solo en 25%. La insuficiencia mitral asoció su severidad con la obstrucción dinámica del tracto de salida del ventrículo izquierdo (p=0,04). Los hipertensos no mostraron mayor hipertrofia ni peor clase funcional (CF). Con una mortalidad global de 8,7%, se observa mejoría sintomática significativa al final del seguimiento (69% vs. 43% ; p=0,001).
Conclusiones: pese al limitado número de pacientes, el RUMHI mostró una población uruguaya con MH de similares características a las descritas en la literatura. La coexistencia con HTA es frecuente (48%), pero no se relacionó con una mayor HVI ni con peor CF. Se detectó búsqueda deficitaria de obstrucción dinámica latente. La población seguida (88,5%) mostró mejoría de la CF, en probable relación con el tratamiento instaurado.
Palabras clave:
MIOCARDIOPATÍA HIPERTRÓFICA
REGISTRO
URUGUAY
Summary
Grounds and objectives: we do not have clinical, morphological, handling or prognostic data of hypertrophic cardiomyopathy (HC) in
Method: we studied a population of 52 patients with HC, with average follow-up of 31.7 ± 12.5 months.
Results: with female predominance (58%) and age 6 to 82 years (50.3± 20.7), 69% present symptoms, prevailing dyspnea (54%) associated with the presence of intraventricular obstruction (p = 0, 048) and syncope, related to a family history of sudden death (p = 0.033). 13% have atrial fibrillation. Twenty-five (48%) associated arterial hypertension (AH); 23 of grade 1. They have typical echocardiographic pattern: small cavity, preserved systolic function, asymmetric left ventricular hypertrophy (LVH) (71%) and maximum thickness on the basal segments of the septum (90%). 58% present dynamic obstruction; latent was searched only in 25%. Mitral regurgitation severity was associated with dynamic left ventricular outflow tract obstruction (p=0,04). Hypertensive subjects showed no major hypertrophy nor worse Functional Class (FC). With a global mortality rate of 8.7%, significant symptomatic improvement was observed at the end of the follow-up (69% vs. 43% ; p=0,001).
Conclusions: despite the limited number of patients, the RUMHI showed a Uruguayan population with HC presenting similar characteristics to those described in literature. Coexistence with AH is frequent (48%) but was not related to a greater LVH nor with worse FC. Insufficient search for latent dynamic obstruction was detected. The population followed (88.5%) showed improvement in FC, likely related to the treatment established.
Key words:
HYPERTROPHIC CARDIOMYOPATHY
REGISTRY
URUGUAY
Introducción
La MH es una miocardiopatía primaria de etiología genética con transmisión autosómica dominante(1,2), ocasionada por mutaciones en genes que codifican diversas proteínas del sarcómero(3). Se caracteriza fenotípicamente por hipertrofia parietal de uno o ambos ventrículos (más frecuentemente el izquierdo), de distribución típicamente asimétrica, sin dilatación ventricular y en ausencia de patología cardíaca o sistémica que pueda explicarla(4). Siendo mayoritariamente familiar, hay casos esporádicos por mutaciones de novo(5,6), luego de las cuales la condición se transmite a la descendencia. Frecuente, pero no invariablemente, se observa obstrucción dinámica del tracto de salida del ventrículo izquierdo(7) (TSVI) ocasionada por la hipertrofia del septum basal anterior junto con un movimiento anómalo de la valva anterior de la válvula mitral que genera a su vez grados variables de insuficiencia valvular(8).
La forma más sencilla y accesible de realizar su diagnóstico es objetivar una HVI sin sobrecarga hemodinámica que la justifique en un ecocardiograma bidimensional(9).
Su prevalencia es de 1/500, según relevamientos ecocardiográficos en poblaciones de individuos de 16 a 87 años pertenecientes a distintas etnias y comunidades(10-12), aunque ciertas características de la enfermedad permiten suponer que podría ser aún mayor(13,14). Si la prevalencia en Uruguay fuera la misma, tendríamos más de 6.500 portadores de MH(15). Se trata de una patología incómoda y desafiante para el cardiólogo clínico, con dificultades diagnósticas derivadas de la amplia prevalencia de hipertensión arterial (HTA) y estenosis aórtica en las poblaciones adultas occidentales y del carácter inconstante y dinámico de la estenosis del TSVI(16). También dificultan su reconocimiento la existencia de una etapa de latencia fenotípica y algunas variantes de distribución de la hipertrofia de difícil detección ecocardiográfica(17,18). Además, las manifestaciones electrocardiográficas pueden llamar a confusión con la cardiopatía isquémica(19).
Adicionalmente, el manejo práctico de los pacientes afectados puede generar áreas de incertidumbre, dadas su heterogeneidad anatómica y clínica, su compleja fisiopatología y la dificultad para estimar el riesgo individual de muerte súbita (MS)(20-23). Se suma su inapropiada percepción como una patología rara, configurando un ámbito proclive a la escasa atención a la enfermedad, el subdiagnóstico y la pobre sistematización de su tratamiento(24).
Durante el año 2009, a instancias de las autoridades de la Sociedad Uruguaya de Cardiología (SUC), conformamos un grupo de trabajo con la finalidad de mejorar el conocimiento y manejo clínico de la MH en Uruguay y decidimos implementar un registro de pacientes con diagnóstico confirmado de la enfermedad. Lo denominamos Registro Uruguayo de Miocardiopatía Hipertrófica (RUMHI) y nos constituimos como Grupo RUMHI.
Objetivos
1. Obtener un mejor conocimiento de las presentaciones clínicas y ecocardiográficas de la enfermedad, y de la forma en que se estudia en nuestro medio.
2. Mantener una base de datos prospectiva para obtener información sobre la evolución sintomática, la realización de nuevos procedimientos terapéuticos y la sobrevida.
3. Explorar la posible asociación del estado sintomático y la mortalidad con diversas variables clínicas y ecocardiográficas, asumiendo que, con el diseño adoptado, la comprobación de una asociación estadísticamente significativa no implica necesariamente una relación de causalidad.
4. Comparar la información obtenida a nivel nacional con la procedente de otros países.
Material y método
Los fundamentos del RUMHI fueron divulgados en una publicación previa(25).
Se trata de un estudio observacional analítico con seguimiento prospectivo (registro activo).
Se incluyeron pacientes cuyo ecocardiograma evidenciara un espesor parietal del ventrículo izquierdo (VI) en diástole ³ 15 mm en cualquier segmento, sin una causa cardíaca o sistémica que la explicara(4), referidos voluntariamente por colegas de todo el país convocados por el Grupo RUMHI. Los niños se incorporaron con un máximo espesor diastólico mayor de 2 desvíos estándar por encima de la media normal para la superficie corporal(26).
Fueron criterios de exclusión del registro:
1. Antecedentes de HTA crónica grado 3 de la European Society of Cardiology (ESC)(27). La HTA grados 1 y 2 no se consideró un criterio de exclusión absoluto si la severidad y la distribución del engrosamiento parietal excedían francamente lo esperable para esa condición.
2. La estenosis valvular aórtica de grado moderado o mayor.
3. Pacientes con diagnóstico de amiloidosis u otros procesos infiltrativos del miocardio.
Se requirió el llenado de un formulario de ingreso con datos clínicos y un formulario de ecocardiografía acompañado de una secuencia dinámica del ecocardiograma o una imagen estática representativa, a ser evaluados por los coordinadores del estudio. Una vez aceptado el caso, se solicitó al colega remitente recabar el consentimiento informado y completar un formulario de inclusión con la totalidad de los datos requeridos por el estudio (figura 1).
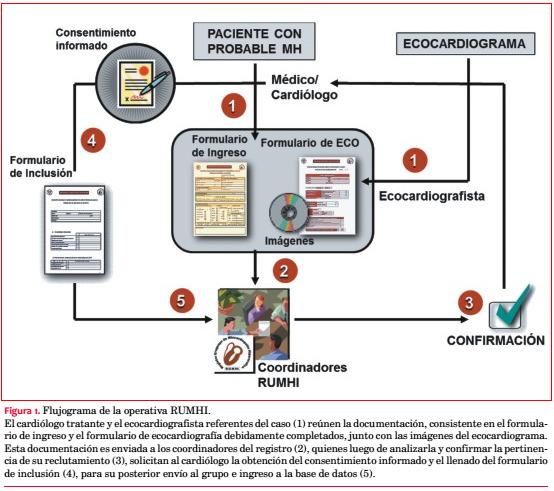
Ingresaron pacientes desde junio de 2010 al 31 de mayo de 2013, registrándose: antecedentes familiares (AF) de MH y de MS, tratamientos invasivos previos, posibles causas de sobrecarga hemodinámica del VI, síntomas, clase funcional (CF), ritmo cardíaco, datos del electrocardiograma (ECG) y datos ecocardiográficos. Se consideró fracción de eyección del VI (FEVI) normal a un valor ³ 55%. Hipertrofia asimétrica del VI implica una relación entre el espesor máximo del septum interventricular (SIV) y el de la pared posterior (PP) ³ 1,5. Un gradiente ³ 30 mmHg en cualquier sector ventricular, basalmente o con maniobras dinámicas se consideró obstrucción.
En el período agosto-noviembre de 2014 los coordinadores realizaron la revisión de historias o establecieron contacto telefónico con los pacientes o sus médicos. Se investigaron tres aspectos: a) sobrevida; b) síntomas y CF, y c) nuevos procedimientos relacionados con la enfermedad.
Estadística
Para el análisis de las variables categóricas se utilizó el test exacto de Fisher (variables dicotómicas) y el test de chi cuadrado de Pearson (variables no dicotómicas). Para la diferencia de medias en las variables continuas se utilizó la prueba no paramétrica de Kruskall Wallis y el test t-Student.
El intervalo de confianza para la media de las variables continuas se obtuvo con un nivel de 95%.
En el análisis del seguimiento se utilizó el test de Mc Nemar para datos nominales pareados.
Para todos los tests se consideró que existe asociación significativa entre las variables si p-valor < 0,05.
Todas las tablas y los gráficos del trabajo se realizaron utilizando el software estadístico R http://www.r-project.org/.
Ética
El protocolo del registro fue diseñado en cumplimiento de los principios de la Declaración de Helsinki de 1975 y sus revisiones posteriores, y recibió la aprobación del Comité de Ética de la SUC y de la Comisión Nacional de Ética en Investigación del Ministerio de Salud Pública.
Resultados
Fueron considerados 59 pacientes; tres se excluyeron por HTA grados 2 o 3 que podía explicar la hipertrofia que presentaban, y cuatro por información insuficiente. Las características clínicas de los 52 incluidos aparecen en la tabla 1; 30 son de sexo femenino (58%).
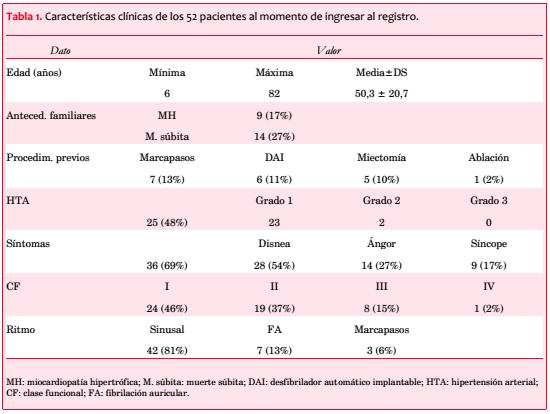
La edad varía entre 6 y 82 años, con media de 50,3 ± 20,7 y distribución bimodal (31 a 40 y 71 a 80 años; figura 2).
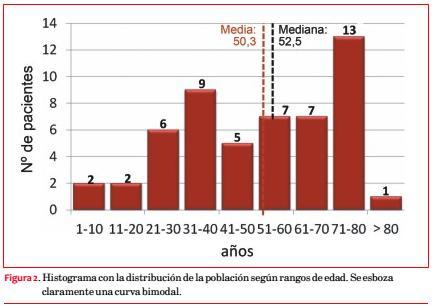
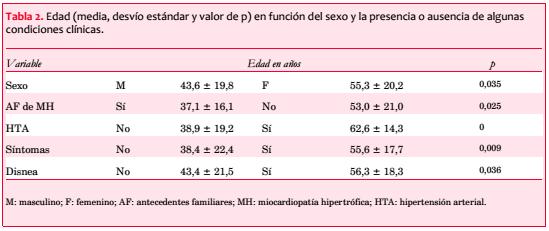
El sexo femenino muestra una edad significativamente mayor (55,3 ± 20,2 vs 43,6 ± 19,8 años; p=0,035). La edad también se asoció con otras características clínicas (tabla 2).
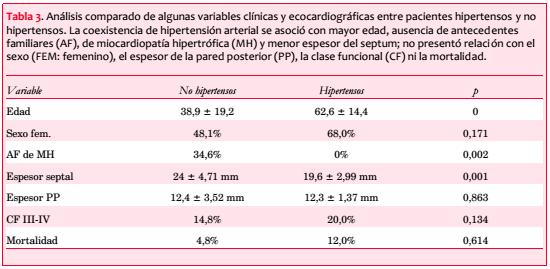
Treinta y seis estaban sintomáticos al momento de su inclusión en el registro (69%), presentando 28 (54%) disnea, 14 (27%) ángor pectoris y 9 (17%) síncope (figura 3). Los pacientes sintomáticos se asociaron con una edad significativamente mayor (55,6 ± 17,7 vs. 38,4 ± 22,4 años; p=0,009). La disnea se asoció a la presencia de obstrucción intraventricular (obstrucción 66,7% vs. no obstrucción 36,4%; p=0,048) y el síncope a los antecedentes familiares de MS (con AF 35,7% vs. sin AF 8,6%; p=0,033). Veinticuatro pacientes (46%) estaban en CF I, 19 (37%) en CF II, 8 (15%) en CF III y 1 (2%) en CF IV, con media de 1,7.
Los pacientes sintomáticos se asociaron con una edad significativamente mayor (p=0,009).
Doce pacientes (23%) tienen dispositivos implantados previamente; 7 (13%) marcapasos definitivo (MPD) y 6 (11%) desfibrilador automático implantable (DAI), incluyendo un paciente con ambos implantes en forma sucesiva, y seis poseen antecedentes de intervenciones de reducción septal: cinco miectomías quirúrgicas (cuatro con sustitución de válvula mitral concomitante) y una ablación septal con alcohol.
El ECG muestra ritmo sinusal en 42 pacientes (81%), fibrilación auricular (FA) en 7 (13%) y 3 tienen ritmo de marcapasos (6%). El complejo QRS se evaluó en 40 pacientes (en 12 individuos no se consignó esta información), resultando anormal en 31 (77,5%). La repolarización ventricular resultó anormal en el 91% de los casos consignados (40 de 44 pacientes).
Valores ecocardiográficos
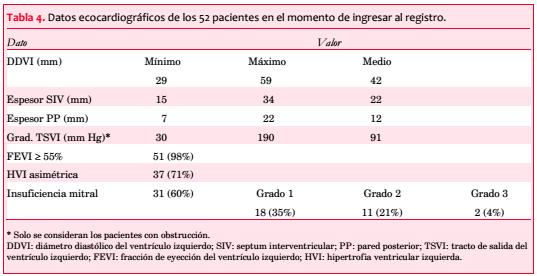
Se observó asimetría de la HVI en 37 pacientes (71%), con máximo espesor del VI en los segmentos basal y medio del SIV en 47 pacientes (90%), en el SIV y el segmento anterobasal en dos, en el SIV y el ápex en uno, en el segmento anteroseptal en otro y en el ápex en un caso (tabla 4).
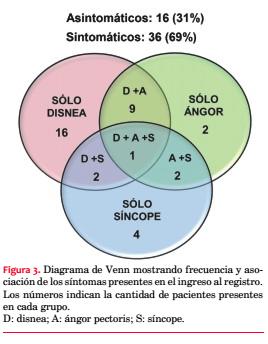
El espesor septal se asoció directa y significativamente con AF de MH (con AF 25,4 ± 1,42; sin AF 21,1 ± 0,67 mm; p=0,009) e inversamente con el antecedente de HTA (hipertensos 19,6 ± 2,99 mm; no hipertensos 24 ± 4,71 mm; p=0,001). El espesor de la pared posterior (PP) no se diferenció según presencia o no de HTA (12,3 ± 1,37 vs 12,4 ± 3,52 mm; p=0,863).
La FEVI resultó normal en 51 pacientes (98%).
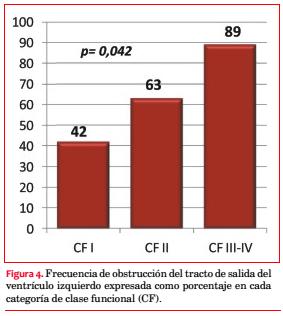
En 30 pacientes (58%) se objetivó obstrucción, pero solo en 13 (25%) se practicaron maniobras para detectar obstrucción latente. La media del gradiente pico registrado fue de 91 mmHg. La obstrucción intraventricular se correlacionó en forma significativa con la CF (p=0,042; figura 4 y tabla 5).
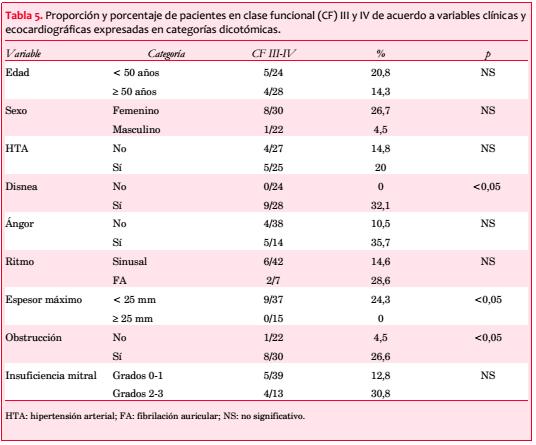
Se observó una paciente de 16 años muy sintomática, con máxima hipertrofia septal media y de ápex y severa estenosis medioventricular, con participación similar del ventrículo derecho (VD) (figura 5). Los demás pacientes presentaron la obstrucción en el TSVI.
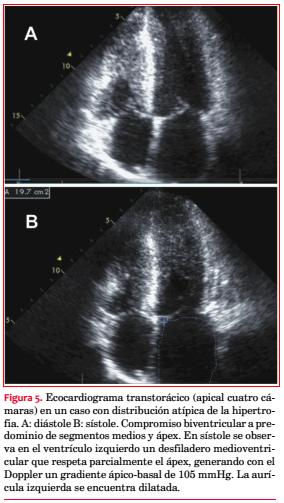
Se comprobó algún grado de insuficiencia mitral (IM) en 31 pacientes (60%): grado 0 (ausente) en 21 (40%); grado 1 (leve) en 18 (35%), grado 2 (moderada) en 11 (21%) y grado 3 (severa) en 2 (4%). Hubo una correlación significativa entre la IM y la obstrucción cuando se la dividió en subgrupos de acuerdo con su severidad (grados 0-1 46,2%; grados 2-3 92,3%, p=0,004; figura 6).
Siete pacientes (13%) tienen una cardiopatía asociada (isquémica en tres, prolapso de válvula mitral en tres, y aneurisma del septum interauricular en uno).
Seguimiento
Fueron seguidos 46 pacientes (88,5%) durante un período de 4 a 50 meses (media de 31,7 ± 12,5 meses). Fallecieron cuatro mujeres: una por accidente cerebrovascular (ACV) de naturaleza no definida; otra por hematoma subdural; otra por insuficiencia cardíaca (IC), y otra por causa no aclarada, aunque el DAI que tenía implantado no registró arritmias agudas previo al deceso. No se encontró correlación significativa de la mortalidad con ninguna de las variables exploradas (tabla 6).
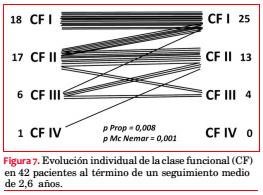
De 42 sobrevivientes reevaluados, 24 (57%) se encuentran asintomáticos y 18 (43%) presentan síntomas: 15 disnea, 1 ángor y 4 episodios sincopales. La comparación de la proporción de sintomáticos al inicio (69%) y al final del seguimiento (43%) muestra una mejoría significativa (p=0,008 para test de igualdad de proporciones; p=0,001 para test de Mc Nemar). La evolución de la CF de los 42 pacientes se muestra en la figura 7.
Durante el seguimiento se indicó implante de DAI en tres pacientes: dos por episodio sincopal con demostración de taquicardia ventricular no sostenida (TVNS) en el Holter y uno por episodio sincopal de causa no aclarada en paciente con antecedentes de MS de cuatro hermanos. Dos fueron implantados; uno no aceptó el procedimiento.
El proyecto original del RUMHI previó el reclutamiento de más de 100 pacientes, expectativa no cumplida a pesar de los esfuerzos realizados en diseño, convocatoria y difusión, ya que dependía de la colaboración de la comunidad cardiológica, que resultó limitada. No obstante tratarse de una población pequeña, el haberse comportado como se describe en la literatura en la mayoría de los aspectos analizados (distribución etaria, rasgos ecocardiográficos, síntoma predominante, correlación positiva entre obstrucción y disnea, frecuencia de FA y mortalidad), permite considerar como probablemente ciertas algunas observaciones adicionales.
La mayoría de individuos con MH son asintomáticos y tienen una expectativa de vida normal(4,28-30). Dos tercios de nuestros pacientes tenían síntomas al ingresar al registro, siendo el más frecuente, como se ha señalado(29), la disnea de esfuerzo, que se presentó en más de la mitad de los casos, seguida por el ángor y en menor proporción por el síncope. Dada la metodología de reclutamiento, nuestra serie proviene de diferentes servicios terciarios, incluyendo pacientes de una policlínica de portadores de MH e individuos que recibieron procedimientos invasivos, y no representa el espectro global de los afectados por esta patología. De acuerdo a informes previos, pueden constituir una subpoblación más sintomática y de mayor riesgo para eventos adversos, incluyendo mortalidad(31-33). La distribución por sexos mostró un definido predominio femenino (58% vs 42%), diferente al equilibrio esperable considerando el patrón hereditario de la enfermedad(34), aunque esto bien puede depender del azar dado el bajo número de pacientes. Como está descrito, las edades abarcaron un amplio espectro, desde individuos en edad pediátrica hasta sujetos añosos, ya que a pesar de la existencia de un período de latencia para el desarrollo de la hipertrofia, la MH puede diagnosticarse a cualquier edad(35). Se observó una configuración bimodal. Diversas características clínicas se distribuyeron en correspondencia con distinta media etaria (tabla 2). En base a ello, sin perder de vista el bajo número de pacientes, ciertas correlaciones permitirían dividir a nuestra población en dos grupos: una cohorte más joven de predominio masculino, con mayor frecuencia de MH detectable en consanguíneos y prevalencia mínima de HTA, con mayor hipertrofia septal aunque con menor frecuencia de síntomas, y otra más añosa con atributos opuestos. Podría especularse en un sustrato genético para explicarlo, ya que diferentes proteínas mutantes pueden determinar divergencias en la edad de aparición de la hipertrofia, su severidad, patrón de distribución y expresión clínica, incluyendo síntomas y riesgo de MS(38-42).
Se han planteado diversos enfoques adicionales para diferenciar la MH de la miocardiopatía hipertensiva, incluyendo el tipo morfológico de la HVI(63), Doppler tisular con strain rate(64-66), estimación con Doppler de la velocidad diastólica en las arterias septales(67), estudios metabólicos con PET y fluordeoxiglucosa(68), centellografía con carnitina y ácidos grasos marcados(69), y tests de regresión de la HVI con tratamiento(70), todos ellos de limitada aplicabilidad clínica. Por el contrario, la RNM se ha demostrado una herramienta de gran utilidad en los diagnósticos positivo y diferencial de la MH, no solo con la cardiopatía hipertensiva, sino con otras miocardiopatías que cursan con aumento del espesor septal(71-73). El realce tardío con gadolinio en los puntos de inserción del VD o su localización mesoparietal en los segmentos de máximo espesor del VI, irían a favor del diagnóstico de MH(74,75).
No está claro si una enfermedad afecta, no ya la expresión morfológica cardíaca, sino también la evolución clínica de la otra. Un estudio poblacional(76) mostró que los portadores de MH sin HTA tenían una mortalidad anual de 0,7%, superponible a la de la población general ajustada por sexo y edad, que ascendía a 5%/año en los hipertensos. En el estudio de Dimitrow y colaboradores(53), la superposición de HTA empeoraba la CF sustancialmente en los pacientes más jóvenes, mientras que entre los mayores la diferencia en CF entre hipertensos y normotensos era insignificante. Por su parte, Karam y colaboradores(51), estudiando una población de 113 pacientes con MH, concluyen que, con la excepción de un mayor engrosamiento parietal, las características clínicas y ecocardiográficas de los pacientes con hipertensión son indistinguibles de las de aquellos de similar edad no hipertensos. Aslam y colaboradores(52), en cambio, no encontraron diferencias significativas ni morfológicas, ni en CF, entre grupos con y sin HTA asociada. De igual modo, los pacientes hipertensos de nuestro registro no mostraron diferencias significativas con los normotensos en su CF ni en la presencia de síntomas (p=0,722).
La prevalencia de FA en nuestra serie (13%), con edad promedio de 50 años, es marcadamente mayor que para un rango etario similar de la población general(77,78) y algo menor que la descrita en la literatura para la MH, en el entorno de 18% a 22%(79-81). La FA en la MH constituye un marcador de riesgo de ACV, discapacidad y mortalidad por insuficiencia cardíaca (IC)(79-83).
Los datos morfológicos y funcionales del ecocardiograma presentan rasgos típicos de la enfermedad.
Como ha sido descrito, en la mayoría de los pacientes la hipertrofia es asimétrica e involucra preferencialmente al SIV en sus segmentos basales(47,48,84,85). Se registró comportamiento obstructivo basal en 54% de los casos, un valor superior al publicado en estudios de previos –25% en la serie de 1.101 pacientes de Maron y colaboradores(86), 27% en los 526 de Autore(87) y 38,5% en los 646 de Casabé y colaboradores(88)–, probablemente por sesgo de inclusión, con elevada proporción de pacientes sintomáticos.
Debemos señalar que en nuestra serie solo se practicaron maniobras sensibilizadoras durante el estudio ecocardiográfico en la cuarta parte de ellos. Solamente en un caso la obstrucción no asentó a nivel del TSVI sino medio ventricular, variante reputada como rara(89-91), que no obstante ocurre hasta en 9,4% en ciertas series(92), y se asocia con mayor discapacidad, disfunción ventricular, ACV y mortalidad súbita y por IC(92,93).
La insuficiencia mitral comparte una base fisiopatológica común con la obstrucción del TSVI y no sorprende que, acorde con informes previos(8), hayamos encontrado una relación directa entre la obstrucción subaórtica y la severidad de la IM concomitante.
Lamentablemente no pudimos contar con datos ecocardiográficos confiables de la dimensión de la aurícula izquierda, otra importante variable pronóstica(95-97).
Se dispuso de trazados electrocardiográficos de adecuada calidad en la mayoría de los pacientes y tanto el complejo QRS como la repolarización ventricular resultaron anormales en 77,5% y 91% de los casos, respectivamente, datos coincidentes con lo registrado en la literatura(98-101).
En tres pacientes hubo asociación con enfermedad coronaria, circunstancia no infrecuente que ha sido señalada en varias publicaciones previas(102-106).
Al cabo de 31,7 meses de seguimiento medio de 46 pacientes, cuatro fallecieron, lo que representa una mortalidad global de 8,7% o anualizada de 3,3%, comparable con series de centros terciarios que reportan una mortalidad de 3% a 6% por año(107,108). Nuevamente el síntoma más frecuente fue la disnea, pero en una proporción menor (36%). El cambio experimentado en la CF se muestra en la figura 7.
Comparando el estado sintomático basal con el evolutivo en los 42 sobrevivientes se observó una diferencia favorable estadísticamente significativa (p=0,001), cuya interpretación podría radicar en un efecto positivo del tratamiento realizado(109), o bien en un sesgo de selección natural por fallecimiento de los más afectados. Sin embargo, de los cuatro pacientes fallecidos, dos estaban al comienzo del estudio en CF I y uno en CF II, y solo uno de ellos falleció por una causa claramente atribuible o relacionada con la enfermedad, como es la IC. Además, el único con antecedentes arrítmicos no mostró arritmias agudas en su DAI.
En tres pacientes surgió la indicación de implante de cardiodesfibrilador automático a causa de su elevado riesgo de MS. Este requerimiento no es contradictorio con la aceptable CF promedio de la población, porque el riesgo de MS en la MH no guarda relación con el grado de limitación funcional, sino que depende de otros factores cuyo valor relativo ha intentado sistematizarse en numerosos trabajos originales y guías de manejo de la enfermedad(4,44,110-115). El brusco e inesperado deceso de individuos jóvenes previamente asintomáticos y de deportistas con estado de salud en apariencia excelente constituye un lamentable recordatorio de que la MS puede ser el primer síntoma de la enfermedad(116-119).
Conclusiones
1) El RUMHI es el primer estudio nacional del perfil clínico-ecocardiográfico de una población con MH, con un seguimiento a mediano plazo del estado sintomático y la sobrevida de los pacientes. Pese al escaso número reclutado, muestra una población uruguaya con MH de similares características a las descritas en otros países: niveles etarios, hipertrofia predominante en segmentos basales del SIV, relación positiva entre la obstrucción dinámica del TSVI y la disnea (síntoma más frecuente) y entre la obstrucción y la IM, elevada frecuencia de FA y mortalidad. Se diferenció en mostrarse mayoritariamente sintomática y con frecuente obstrucción dinámica del tracto de salida, probablemente por sesgo de inclusión, observándose el síncope particularmente en aquellos pacientes con AF de MS.
2) La coexistencia con HTA es frecuente (48%), pero esta asociación no se relacionó con una mayor hipertrofia ventricular ni con peor CF, reforzando el concepto de que no debe descartarse la presencia de MH solo por el antecedente de HTA.
3) En los estudios ecocardiográficos se detectaron deficiencias en la búsqueda de obstrucción dinámica latente.
4) La población seguida (88,5%) mostró mejoría de la CF, en probable relación con el tratamiento instaurado.
Bibliografía
1. Maron B, Towbin J A, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies. An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807-1816.
2. Watkins H, Ashrafian H, McKenna W. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart 2008; 94: 1264-1268.
3. Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol 2010; 25: 205-209.
4. Gersh B, Maron B, Bonow R, Dearani J, Fifer M, Link M, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy. A Report of the
5. Watkins H, Thierfelder L, Hwang D, McKenna W, Seidman J, Seidman C. Sporadic hypertrophic cardiomyopathy due to de novo myosin mutations. J Clin Invest 1992; 90: 1666-1671.
6. Olson T, Doan T, Kishimoto N, Whitby F, Ackerman M, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell cardiol 2000; 32: 1687-1694.
7. Maron M, Olivotto I, Zenovich A, Link M, Pandian N, Kuvin J, et al. Hypertrophic cardiomyopathy is predominantly a disease o left ventricular outflow tract obstruction. Circulation 2006; 114: 2232-2239.
8. Yu E H, Omran A, Wigle E, Williams W, Siu S, Rakowsky H. Mitral regurgitation in hypertrophic obstructive cardiomyopathy: relation with obstruction and relief with myectomy. J Am Coll Cardiol 2000; 36: 2219-2225.
9. Williams L, Frenneaux P, Steeds R. Echocardiography in hypertrophic cardiomyopathy diagnosis, prognosis and role in management. Eur J Echocardiogr 2009; 10: 9-14
10. Maron B, Gardin J, Flack J, Gidding S, Kurosaki T, Bild D. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Circulation 1995; 92: 785-789.
11. Maron B, Mathenge R, Casey S, Poliac L, Longe T. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J Am Coll Cardiol 1999, 33: 1590-1595.
12. Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: A population-based echocardiographic analysis 0f 8080 adults. The Am J Medicine 2004; 116: 14-18.
13. Fananapazir L, Epstein N. Prevalence of hypertrophic cardiomyopathy and limitations of screening methods. Circulation 1995; 92: 700-704.
14. Semsarian C, Ingles J, GenCouns D, Maron M, Maron B. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65: 1249-1254.
15. Instituto Nacional de Estadística, Uruguay. Resultados del Censo de Población 2011: población, crecimiento y estructura por sexo y edad. [Internet]. Uruguay: INE; 2011 [consultado 4 Nov 2015]. Disponible en: http://www.ine.gub.uy
16. Wigle ED. The diagnosis of hypertrophic cardiomyopathy. Heart 2001; 86: 709-714.
17. Rickers M, Wilker N, Jerosh-Herold M, Casey S, Panse P, Panse N, et al. Utility of cardiac magnetic resonance imaging in the diagnostic of hypertrophic cardiomyopathy. Circulation 2005; 112: 855-861.
18. Maron M, Maron B, Harrigan C, Buros J, Gibson C, Olivotto I, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol 2009; 54: 220-228.
19. Dumont C, Monserrat L, Soler R, Rodríguez E, Fernández X, Peteiro J, et al. Interpretation of electrocardiographic abnormalities in hypertrophic cardiomyopathy with Cardiac magnetic resonance. Eur Heart J 2006; 27: 1725-1731.
21. Maron B. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation 2010; 121: 445-456.
22. Spirito P, Autore C, Formisano F, Egidy Assenza G, Biagini E, Haas T, et al. Risk of sudden death and outcome in patients with hypertrophic cardiomyopathy with benign presentation and without risk factors. Am J cardiol 2014; 113: 1550-1555.
23. Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron ES. Independent Assessment of the European Society of Cardiology Sudden Death Risk Model for Hypertrophic Cardiomyopathy. Am J Cardiol. 2015 Sep 1; 116(5):757-64. doi: 10.1016/j. amjcard.2015.05.047. Epub 2015 Jun 4
24. Olivotto I, Cecchi F. The epidemiologic evolution and present perception of hypertrophic cardiomyopathy. Ital Heart J 2003; 4: 596-601.
25. Estigarribia J, Vidal I, Báez A, Vidal L. Miocardiopatía hipertrófica. Aspectos conceptuales de la enfermedad y fundamentos del Registro Uruguayo de Miocardiopatía Hipertrófica. Rev Urug Cardiol 2011; 26: 27-37.
26. Kampmann C, Wiethoff CM, Wenzel A, Stolz G, Betancor M, Wippermann CF, et al. Normal values of M mode echocardiographic measures of more than 2000 healthy infants and children in Central Europe. Heart 2000; 83: 667-672.
27. Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Böhm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension. The task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34: 2159-2219.
28. Takagi E, Yamakado T, Nakano T. Prognosis of completely asymptomatic adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33: 206-11.
29. Maron B, Casey S, Poliac L, Gohman T, Almquist A, Aeppli D. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA 1999; 281: 650-655.
30. Maron B, Casey S, Haas T, Kitner C, Garberich R, Lesser J. Hypertrophic cardiomyopathy with longevity to 90 years or older. Am J Cardiol 2012; 109: 1341-1347.
31. Spirito P, Chiarella F, Carratino L, Zoni Berisso M, Belloti P, Vecchio C. Clinical course and prognosis of hypertrophic cardiomyopathy in an outpatient population. N Engl J Med 1989; 320: 749-755.
32. Sorajja P, Nishimura R, Gersh B, Dearani JA, Hodge D, Wiste H, et al. Outcome of mildly symptomatic or asymptomatic obstructive hypertrophic cardiomyopathy: A long-term follow-up study. J Am Coll Cardiol 2009; 54: 234-241.
33. Enriquez A, Goldman M. Management of hypertrophic cardiomyopathy. Ann Global Health 2014; 80: 35-45.
34. Ho C Y. Genetic considerations in hypertrophic cardiomyopathy. Progr Cardiovasc Dis 2012; 54: 456-60.
35. Wilkinson J, Sleeper A, Álvarez J, Bublik N, Lipshultz S, and the pediatric cardiomyopathy study group. The Pediatric Cardiomyopathy Registry: 1995-2007. Progr Pediatr Cardiol 2008; 25: 31-36.
36. Moak JP, Kaski JP. Hypertrophic cardiomyopathy in children. Heart 2012; 98: 1044-1054.
37. Kubo T, Kitaoka H, Okawa M, Nishinaga M, Doi Y. Hypertrophic cardiomyopathy in the elderly. Geriatr Gerontol 2010; 10: 9-16.
38. Niimura H, Patton K, McKenna W, Soults J, Maron B, Seidman J G et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002; 105: 446-451.
39. McKenna W, Coccolo F, Elliot P. Genes and disease expression in hypertrophic cardiomyopathy. Commentary. The Lancet 1998; 352: 1162-1163.
40. Varnava A, Elliot P, Baboonian C, Davidson F, Davies M, McKenna W. Hypertrophic cardiomyopathy. Histopathological features of sudden death in cardiac troponin T disease. Circulation 2001; 104: 1380-1384.
41. Chung MW, Tsoutsman T, Semsarian C. Hypertrophic cardiomyopathy: from gene defect to clinical disease. Cell Res 2003; 13: 9-20.
42. Maron B, Rowin E, Casey S, Haas T, Chan R, Udelson L, et al. Risk stratification and outcome of patients with hypertrophic cardiomyopathy ³60 years of age. Circulation 2013; 127: 585-593.
43. Spirito P, Autore C, Rapezzi C, Bernabó P, Badagliacca R, Maron M, et al. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation 2009; 119: 1703-1710.
44. Elliot P, Anastasakis A, Borger M, Borggrefe M, Cecchi F, Charron P, et al. Guía de práctica clínica de la ESC 2014 sobre el diagnóstico y manejo de la miocardiopatía hipertrófica. Grupo de trabajo de la ESC para el diagnóstico y manejo de la miocardiopatía hipertrófica. Rev Esp Cardiol 2015; 68: 63.e1-e52.
45. Ho Hee-Hwa, Lee K, Lau Chu-Pak, Tse Hung-Fat. Clinical characteristics of and long-term outcome in chinese patients with hypertrophic cardiomyopathy. Am J Med 2004; 116: 19-23.
46. Carasso S, Yang H, Woo A, Jamorski M, Wigle E, Rakowski H. Diastolic myocardial mechanics in hypertrophic cardiomyopathy. J Am Soc Echocardiogr 2010; 23: 164-171.
47. Klues H, Schiffers A, Maron B. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll cardiol 1995; 26: 1699-1708.
48. Nagueh S, Mahmarian J. Noninvasive cardiac imaging in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2006; 48: 2410-2422.
49. Arias A, Bagnati R, Pérez de Arenaza D, Oberti P, Falconi M, Pizarro R, et al. Perfil clínico de pacientes con miocardiopatía hipertrófica en un hospital universitario. Rev Argent Cardiol 2014; 82:366-372.
50. Yoshinaga M, Yoshikawa D, Ishii H, Irashiki A, Okumura T, Kubota A, et al. Clinical characteristics and long-term outcome of hypertrophic cardiomyopathy. Results from the Aichi hypertrophic cardiomyopathy (AHC) registry. Int Heart J 2015; 56: 415-420.
51. Karam R, Lever H, Healy B. Hypertensive hypertrophic cardiomyopathy or hypertrophic cardiomyopathy with hypertension?: A study with 78 patients. J Am Coll Cardiol 1989; 13: 580-584.
52. Aslam F, Haque A, Foody J A, Shirani J. The frequency and functional impact of overlapping hypertension on hypertrophic cardiomyopathy: a single-center experience. J Clin Hypertens (
53. Dimitrow P, Czarnecka D, Kawecka-Jaszcz K, Dubiel JS. The frequency and functional impact of hypertension overlapping on hypertrophic cardiomyopathy: comparison between older and younger patients. J Human Hypertension 1998; 12: 633-634.
54. Ministerio de Salud Pública. 2a Encuesta nacional de factores de riesgo de enfermedades crónicas no transmisibles [Internet]. 2015 [consultado 4 Nov 2015]. Montevideo: MSP. Disponible en: http://www.msp.gub.uy
55. Alcalai R, Seidman J, Seidman C. Genetic basis of hypertrophic cardiomyopathy. From bench to the clinics. J Cardiovasc Electrophysiol 2008; 19: 104-110.
56. Keren A, Syrris P, McKenna W. Hypertrophic cardiomyopathy: the genetics determinants of clinical disease expression. Nature Clin Pract Cardiovasc Med 2008; 5: 158-168.
57. Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol 2002; 17: 242-252.
58. Tarazi RC,
59. Bella JN, Göring H. Genetic epidemiology of left ventricular hypertrophy. Am J Cardiovasc Dis 2012; 2: 267-278.
60. Devereux RB, Alonso DR, Lutas EM, Pickering TG,
61. Desai M, Ommen S, McKenna W, Lever H, Elliott P. Imaging phenotype versus genotype in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging 2011; 4: 156-168.
62. Syed IS, Ommen S, Breen J, Tajik AJ. Hypertrophic cardiomyopathy: Identification of morphological subtypes by echocardiography and cardiac magnetic resonance imaging. Imaging vignette. J Am Coll Cardiol Img 2008; 1: 377-379.
63. Binder J, Ommen S, Gersh B, Van Driest S, Tajik AJ, Nishimura R, et al. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc 2006; 81: 459-467.
64. Kato T, Noda A, Izawa H, Yamada A, Obata K, Nagata K, et al. Discrimination of nonobstructive hypertrophic cardiomyopathy from hypertensive left ventricular hypertrophy on the basis of strain rate imaging by tissue Doppler ultrasonography. Circulation 2004; 110: 3808-3814.
65. Serri K, Reant P, Lafitte M, Berhouet M, Le Bouffos V, Roudaut R, et al. Global and regional myocardial function quantification by two-dimensional strain. J Am Coll Cardiol 2006; 47: 1175-1181.
66. Baratta S, Chejtman D, Fernández H, Ferroni F, Bilbao J, Kotliar C, et al. Valor clínico de la utilización del strain rate sistólico en el estudio de distintas formas de hipertrofia ventricular izquierda. Rev Argent Cardiol 2007; 75: 367-373
67. Sherrid M, Mahenthiran J, Casteneda V, Fincke R, Gasser M, Barac I, et al. Comparison of diastolic septal perforator flow velocities in hypertrophic cardiomyopathy versus hypertensive left ventricular hypertrophy. Am J Cardiol 2006; 97: 106-112.
68. Shiba N, Kagaya Y, Ishide N, Takeyama D, Yamane Y, Chida M, et al. Myocardial glucose metabolism is different between hypertrophic cardiomyopathy and hypertensive heart disease associated with asymmetrical septal hypertrophy. Tohoku J Exp Med 1997; 182: 125-138.
69. Nakamura T, Sugihara H, Kinoshita N, Yoneyama S, Azuma A, Nakagawa M. Can serum carnitine levels distinguish hypertrophic cardiomyopathy from hypertensive hearts? Hypertension 2000; 36: 215-219.
70. Takeda A, Takeda N. Different pathophysiology of cardiac hypertrophy in hypertension and hypertrophic cardiomyopathy. J Mol Cell Cardiol 1997; 29: 2961-2965.
71. Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part 2, Differential diagnosis, risk stratification and posttreatment MRI appearances. Am J Roentgenol. 2007; 189: 1344-1352.
72. To AC, Dhillon A, Desai M. Cardiac magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol Img 2011; 4: 1123-1137.
73. Maron B, Maron M. Clinical impact of contemporary cardiovascular magnetic resonance imaging in hypertrophic cardiomyopathy. Circulation 2015; 132: 292-298.
74. Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part 1, MRI appearances. Am J Roentgenol. 2007; 189: 1335-1343.
75. Noureldin R, Liu S, Nacif M, Judge D, Halushka M, Abraham T, et al. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J Cardiovasc Mag Res 2012; 14: 17-30.
76. Cannan C, Reeder G, Bailey K, Melton III L J, Gersh B. Natural history of hypertrophic cardiomyopathy. A population-based study, 1976 through 1990. Circulation 1995; 92: 2488-2495.
77. Go AS, Hylek E, Phillips K Chang Y, Henault L, Selby J, et al. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: the AnTicoagulation and Risk factors In Atrial fibrillation (ATRIA) study. JAMA 2001; 285: 2370-2375.
78. Wilke T, Groth A, Mueller S, Pfannkuche M, Verheyen F, Linder R, et al. Incidence and prevalence of atrial fibrillation: an analysis based on 8.3 million patients. Europace 2013; 15: 486-493.
79. Higashikawa M, Nakamura Y, Yoshida M, Kinoshita M. Incidence of ischemic strokes in hypertrophic cardiomyopathy is markedly increased if complicated by atrial fibrillation. Jpn Circ J 1997; 61: 673-681.
80. Siontis K, Geske J, Ong K, Nishimura R, Ommen S, Gersh B. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical correlations and mortality in a large high risk population. J Am Heart Assoc. 2014; 3:e00 1002 doi: 10. 1161/ JAHA. 114. 001002.
81. Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014 Mar; 100(6):465-72. doi: 10.1136/heartjnl-2013-304276. Epub 2013 Sep 7.
82. Olivotto I, Cecchi F, Casey S, Dolara A, Traverse J, Maron B. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104: 2517-1524.
83. Maron B, Olivotto I, Bellone P, Conte MR, Cecchi F, Flygenring B. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39: 301-307.
84. Losi MA, Nistri S, Galderisi M, Betocchi S, Cecchi F, Olivotto I, et al. Echocardiography in patients with hypertrophic cardiomyopathy: usefulness of old and new techniques in the diagnosis and the pathophysiological assessment. Cardiovasc Ultrasound 2010; 8: 7-26.
85. Afonso L, Bernal J, Bax JJ, Abraham T. Echocardiography in hypertrophic cardiomyopathy: The role of conventional and emerging technologies. J Am Coll Cardiol Img 2008; 1: 787-800.
86. Maron M, Olivotto I, Betocchi S, Casey S, Lesser J, Losi MA. Effect of left ventricular outflow tract obstruction on clinical outcome of hypertrophic cardiomyopathy. New Engl J Med 2003; 348: 295- 303.
87. Autore C, Bernabó P, Barillá C, Bruzzi P, Spirito P. The prognostic importance of left ventricular outflow obstruction in hypertrophic cardiomyopathy varies in relation to severity of symptoms. J Am Coll Cardiol 2005; 45: 1076-1080.
88. Casabé JH, Fernández A, Renedo MF, Guevara E, Favaloro LE, Favaloro R. Endocarditis infecciosa en la miocardiopatía hipertrófica. Rev Argent Cardiol [Internet]. 2015 Ago [consultado 4 Nov 2015]; 83(4): [aprox.3p.]. Disponible en: http://dx.doi.org/10.7775/rac.es.v83.i4.5476.
89. de Almeida Gripp E, Garcia M, do Amaral SI, Rabischoffsky R, Pupo Barbosa F, Riskalla Correa R, et al. Miocardiopatía hipertrófica medioventricular asociada a aneurisma apical: Estudio ecocardiográfico de dos casos. Rev Bras Ecocardiogr Imagem Cardiovasc. 2012; 25: 236-239.
90. Petrou E, Kyrzopoulos S, Sbarouni E, Tsiapras D, Voudris V. Mid-ventricular hypertrophic obstructive cardiomyopathy complicated by an apical aneurysm, presenting as ventricular tachycardia. J Cardiovasc Ultrasound 2014; 22: 158-159.
91. Cianciulli TF, Saccheri MC, Konopka I, Serans D, Acunzo R, García Escudero A, et al. Subaortic and mid-ventricular obstructive hypertrophic cardiomyopathy with apical aneurysm: a case report. Cardiovascular Ultrasound 2006; 4: 15-20.
92. Minami Y, Kajimoto K, Terajima Y, Yashiro B, Okayama D, Haruki S, et al. Clinical implications of midventricular obstruction in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2011; 57: 2346-2355.
93. Efthimiadis GK, Pagourelias ED, Parcharidou D, Gossios T, Kamperidis V, Theofilogiannakos EK, et al. Clinical characteristics and natural history of hypertrophic cardiomyopathy with midventricular obstruction. Circ J 2013; 77:2366–2374.
94. Maron B, Maron J, Wigle E, Braunwald E. The 50-year history, controversy and clinical implications of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy: From idiopathic hypertrophic subaortic stenosis to hypertrophic cardiomyopathy. J Am Coll Cardiol 2009; 54: 191-200.
95. Nistri S, Olivotto I, Betocchi S, Losi MA, Valsecchi G, Pinamonti B, et al. Prognostic significance of left atrial size in patients with hypertrophic cardiomyopathy (from the Italian Registry for Hypertrophic Cardiomyopathy). Am J Cardiol 2006; 98: 960–965.
96. Yang Woo-Ing, Shim CI, Kim YJ, Kim Sung-Ai, Rhee SJ, Choi Eui-Young, et al. Left atrial volume index: A predictor of adverse outcome in patients with hypertrophic cardiomyopathy. J Am Soc Echocardiogr 2009; 22: 1338-1343.
97. Losi MA, Betocchi S, Barbati G, Parisi V, Tocchetti C, Pastore F, et al. Prognostic significance of left atrial volume dilatation in patients with hypertrophic cardiomyopathy. J Am Soc Echocardiogr 2009; 22: 76-81.
98. Savage D, Seides SF, Clark C, Henry W, Maron B, Robinson F, et al. Electrocardiographic findings in patients with obstructive and nonobstructive hypertrophic cardiomyopathy. Circulation 1978; 58: 402-408.
99. Maron B. The electrocardiogram as a diagnostic tool for hypertrophic cardiomyopathy: Revisited. Ann Noninvasive Electrocardiol. 2001; 6: 277-279.
100. Konno T, Shimizu M, Ino H, Yamaguchi M, Terai H, Uchiyama K, et al. Diagnostic value of abnormal Q waves for identification of preclinical carriers of hypertrophic cardiomyopathy based on a molecular genetic diagnosis. Eur Heart J 2004; 25: 246-251.
101. Lakdawala N, Thune JJ, Maron B, Cirino A, Havndrup O, Bundgaard H, et al. Electrocardiographic features of sarcomere mutations carriers with versus without clinically overt hypertrophic cardiomyopathy. Am J Cardiol 2011; 108: 1606-1613.
102. Walston A, Behar BS. Spectrum of coronary heart disease in idiopatic hypertrophic subaortic stenosis. Am J Cardiol 1976; 38: 12-16.
103. Cokkinos DV, Krajcer Z, Leachman RD. Coronary artery disease in hypertrophic cardiomyopathy. Am J Cardiol 1985; 55: 1437-8.
104. Sorajja P, Ommen S, Nishimura R, Gersh B, Berger P, Tajik A. Adverse prognosis in patients with hypertrophic cardiomyopathy who have epicardial coronary artery disease. Circulation 2003; 108: 2342-2348.
105. Soca G, Cura L, Genta F, Montero H, Ligüera L, Dayan V, et al. Evaluación de resultados quirúrgicos de la miectomía como tratamiento de la miocardiopatía hipertrófica obstructiva. Experiencia de un centro en Uruguay. Rev Urug Cardiol 2010; 25: 5-10.
106. Estigarribia J. Miocardiopatía hipertrófica y cardiopatía isquémica. Una asociación frecuente. Rev Urug Cardiol 2010; 25: 229-234.
107. Maron B. Hypertrophic cardiomyopathy. A systematic review. JAMA 2002; 287: 1308-1320.
108. Hecht GM, Maron B. Conceptos actuales en miocardiopatía hipertrófica. Rev Argent Cardiol 2003; 71: 446-452.
109. Maron BJ, Rowin EJ, Casey SA, Link MS, Lesser JR, Chan RH et al. Hypertrophic Cardiomyopathy in Adulthood Associated With Low Cardiovascular Mortality With Contemporary Management Strategies. J Am Coll Cardiol. 2015 May 12; 65(18):1915-28. doi: 10.1016/j.jacc.2015.02.061.
110. Saumarez R, Pytkowsky M, Sterlinski M, Bourke J, Clague J, Cobbe S, et al. Paced ventricular electrogram fractionation predicts sudden cardiac death in hypertrophic cardiomyopathy. Eur Heart J 2008; 29: 1653-1661.
111. Östman-Smith I, Wisten A, Nylander E, Bratt E, de Wahl-Granelli A, Oulhaj A, et al. Electrocardiographic amplitudes: a new risk factor for sudden death in hypertrophic cardiomyopathy. Eur Heart J 2010; 31: 439-449.
112. Dimitrow P, Chojnowska L, Rudzinski T, Piotrowski W, Ziotkowska L, Wojtarowicz A, et al. Sudden death in hypertrophic cardiomyopathy: old risk factors re-assessed in a new model of maximalized follow-up. Eur J Cardiol 2010; 31. 3084-3093.
113. Miller M, Gomes J, Fuster V. Risk stratification of sudden cardiac death in hypertrophic cardiomyopathy. Nature Clin Pract Cardiovasc Med 2007; 4: 667-676
114. Adabag S, Maron B, Appelbaum E, Harrigan C, Buros J, Gibson M, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol 2008; 51: 1369-1374.
115. O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur Heart J 2014; 35: 2010-2020 doi: 10.1093/eurheartj/eht439.
116. Maron B. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin 2007; 25: 399-414. doi:10.1016/j.ccl.2007.07.006
117. Rowland T. Sudden unexpected death in young athletes: reconsidering “Hypertrophic Cardiomyopathy”. Pediatrics 2009; 123: 1217-1222.
118. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998; 339: 364-369.
119. Panhuyzen-Goedkoop N, Verheugt F. Sudden cardiac death due to hypertrophic cardiomyopathy can be reduced by pre-participation cardiovascular screening in young athletes. Editorial. Eur Heart J 2006; 27: 2152-2153.
Apéndice
Impulsados por las autoridades de la SUC del año 2009, se realizaron reiterados llamados abiertos a través de la lista de correos electrónicos de socios para incorporar a todos los colegas interesados en trabajar en el proyecto. Estos fueron, en orden alfabético, los Dres. Álvaro Báez, Eduardo Benkel, Alejandro Cuesta, Aída De Luca, Gabriel Echegaray, Natalia Esmite, Jorge Estigarribia, Bettina Frugoni, Mónica Giambruno, Alejandra Machado, Cristina Martínez, Silvia Mato, Carlos Peluffo, José Pérez, Nelson Pizzano, Edgardo Sandoya, Inés Vidal, Luis Vidal y Fernando Wajner.
En agosto de 2010, fueron designados como coordinadores del Registro los Dres. Álvaro Báez, Jorge Estigarribia, Inés Vidal y Luis Vidal, siendo designada la Dra. Inés Vidal como investigadora responsable ante la Comisión Nacional de Ética en Investigación. Los Dres. Álvaro Báez e Inés Vidal, en su condición de ecocardiografistas, fueron además los encargados, en caso de duda de cumplimiento con los criterios de inclusión al Registro, de revisar el ecocardiograma presentado.
Los colegas que remitieron casos finalmente aceptados e incluidos fueron designados médicos colaboradores; la lista completa por orden alfabético es la siguiente:
Beatriz Ansín, Álvaro Báez, Gerard Burdiat, Aída De Luca, Guillermo Dermit, Jorge Estigarribia, Mónica Giambruno, Ricardo Lorenzo, María Alejandra Machado, Ruben Madera, Florencia Maglione, Silvia Mato, Félix Rivedieu, Marcelo Santoro, Alicia Torterolo, Horacio Vázquez, Inés Vidal y Luis Vidal.

