










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Uruguaya de Cardiología
versión On-line ISSN 1688-0420
Rev.Urug.Cardiol. vol.30 no.2 Montevideo ago. 2015
Artículo seleccionado
Almanac 2014: las Revistas de las Sociedades Nacionales presentan investigaciones seleccionadas que han determinado recientes avances en la cardiología clínica
Miocardiopatías
Oliver P Guttmann1, Saidi A Mohiddin2, Perry M Elliott1
Este artículo fue publicado en Heart 2014;100:756-64. doi:10.1136/heartjnl-2013-305420, y es reproducido y traducido con autorización
1. Unidad de Cardiopatías Hereditarias, The Heart Hospital, University College London, Londres, Reino Unido.
2. Departamento de Cardiología, The London Chest Hospital, Londres, Reino Unido.
Correspondencia: Profesor Perry M. Elliott, Unidad de Cardiopatías Hereditarias, The Heart Hospital, University College London, 16-18 Westmoreland Street, London W1G8PH, Londres, Reino Unido. Correo electrónico: perry.elliott@ucl.ac.uk
Recibido: 16 de diciembre de 2013; aceptado: 5 de febrero de 2014; publicado en línea por primera vez: 6 de marzo de 2014.
Resumen
Las miocardiopatías son trastornos miocárdicos que no se pueden explicar por condiciones de carga anormales o enfermedad coronaria. Se clasifican en una serie de fenotipos morfológicos y funcionales de etiología genética y no genética. Los temas dominantes en los trabajos publicados en 2012-2013 son similares a los publicados en Almanac 2011, como el uso (y la interpretación) de las pruebas genéticas, el desarrollo y la aplicación de nuevas técnicas imagenológicas no invasivas, y el uso de biomarcadores séricos para el diagnóstico y el pronóstico. Una importante innovación desde el último número de Almanac es la aparición de modelos más sofisticados de predicción de eventos clínicos adversos.
Introducción
Las miocardiopatías son trastornos miocárdicos que no se pueden explicar por condiciones de carga anormales o enfermedad coronaria. Se las clasifica en una serie de fenotipos morfológicos y funcionales que pueden ser causados por mecanismos genéticos y no genéticos. Los temas dominantes en los trabajos publicados en 2012-2013 son similares a los comunicados en Almanac 2011, como el uso (y la interpretación) de las pruebas genéticas, el desarrollo y la aplicación de nuevas técnicas imagenológicas no invasivas y el uso de biomarcadores séricos para diagnóstico y pronóstico. Una importante innovación desde el último número de Almanac es la aparición de modelos más sofisticados de predicción de eventos clínicos adversos.
Miocardiopatía hipertrófica
Imagenología cardíaca y biomarcadores circulantes
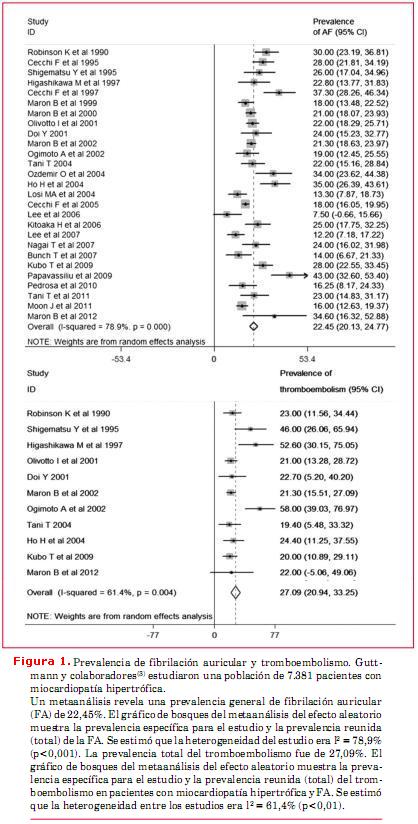
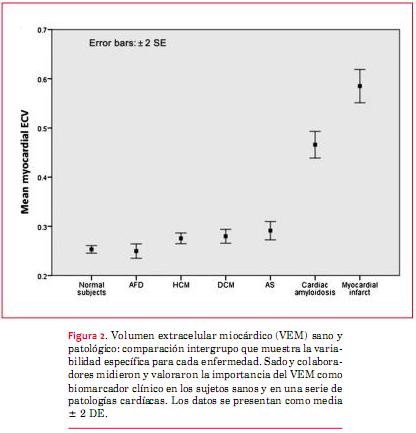
La miocardiopatía hipertrófica (MCH) se presenta en uno de cada 500 adultos y en la mayoría de los individuos es heredada como un rasgo autosómico dominante provocado por mutaciones en los genes de proteínas sarcoméricas cardíacas y está asociada a un mayor riesgo de muerte súbita cardíaca (MSC), disfunción ventricular progresiva y accidente cerebrovascular (ACV) (figura 1)(1-3). Si bien hay herramientas diagnósticas como el electrocardiograma (ECG) y el ecocardiograma, que siguen siendo fundamentales para el diagnóstico y tratamiento de la MCH, la resonancia magnética cardíaca (RMC) mejora la precisión diagnóstica y aporta información fenotípica adicional en pacientes con enfermedad ya establecida (figura 2)(4-7). Por ejemplo, en un estudio, la RMC identificó hipertrofia en aproximadamente un 10% de los portadores de mutación sarcomérica cuyo ecocardiograma mostraba un espesor parietal normal(8). Hay nuevas secuencias de RMC, como el T1 mapping, que brindan una estimación cuantitativa del volumen extracelular del miocardio (VEM) y, por lo tanto, son una medida sucedánea de fibrosis intersticial(5). En un estudio se reportó el aumento del VEM en individuos con mutaciones sarcoméricas pero sin hipertrofia del ventrículo izquierdo (VI)(9). Estos hallazgos sugieren que el uso selectivo de la RMC puede ser de utilidad en el screening familiar, particularmente cuando hay otras características congruentes con MCH, como anomalías del ECG.
Numerosos trabajos han investigado los biomarcadores como herramienta diagnóstica y pronóstica, y han mostrado predecir un pobre pronóstico en pacientes con insuficiencia cardíaca(12). En un estudio de 772 pacientes con MCH, el péptido natriurético cerebral (BNP, por su sigla en inglés) se comportó como un predictor independiente de morbilidad y mortalidad(13). En otro estudio de 183 pacientes ambulatorios estables, el NT-pro BNP plasmático resultó predictor de eventos relacionados con insuficiencia cardíaca(14) y fue predictor de muerte relacionada con el trasplante e insuficiencia cardíaca, pero no de muerte súbita o descargas inapropiadas del cardiodesfibrilador implantable (CDI)(15). Otro estudio de 183 pacientes comunica concentraciones séricas elevadas de troponina T cardíaca de alta sensibilidad como predictor de resultados adversos en la MCH(16).
Estrategias de tratamiento
Actualmente, el manejo de los individuos con MCH apunta a la prevención de la MSC y el ACV, el alivio de los síntomas asociados a la obstrucción del tracto de salida del ventrículo izquierdo (OTSVI) refractarios a la medicación, y la paliación de los síntomas limitantes provocados por disfunción sistólica o diastólica. Desde el último número de Almanac no ha habido muchos avances en la terapia, pero se ha demostrado que la terapia profiláctica temprana con betabloqueantes en los pacientes físicamente activos (NYHA I-II) con OTSVI provocable, logra reducir los gradientes del tracto de salida durante el ejercicio fisiológico(17). Otro estudio ha confirmado el beneficio adicional de la disopiramida en la terapia de los pacientes sintomáticos con obstrucción resistente a la terapia inicial con betabloqueantes o verapamil(18).
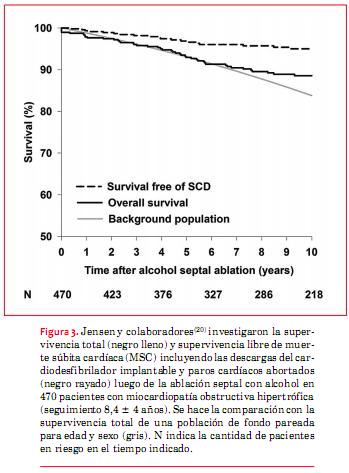
Se ha vuelto a examinar la eficacia de la estimulación bicameral para el tratamiento de la OTSVI sintomática refractaria(24,25). En una reciente revisión de Cochrane se hizo evidente que todos los datos derivan de estudios pequeños y que los pocos ensayos aleatorizados(26,27) se concentran en medidas de resultados fisiológicos y no en la evaluación de variables clínicas “duras”. Por consiguiente, el estudio recomienda realizar ensayos de mayor tamaño y alta calidad(28).
Prevención de la muerte súbita cardíaca
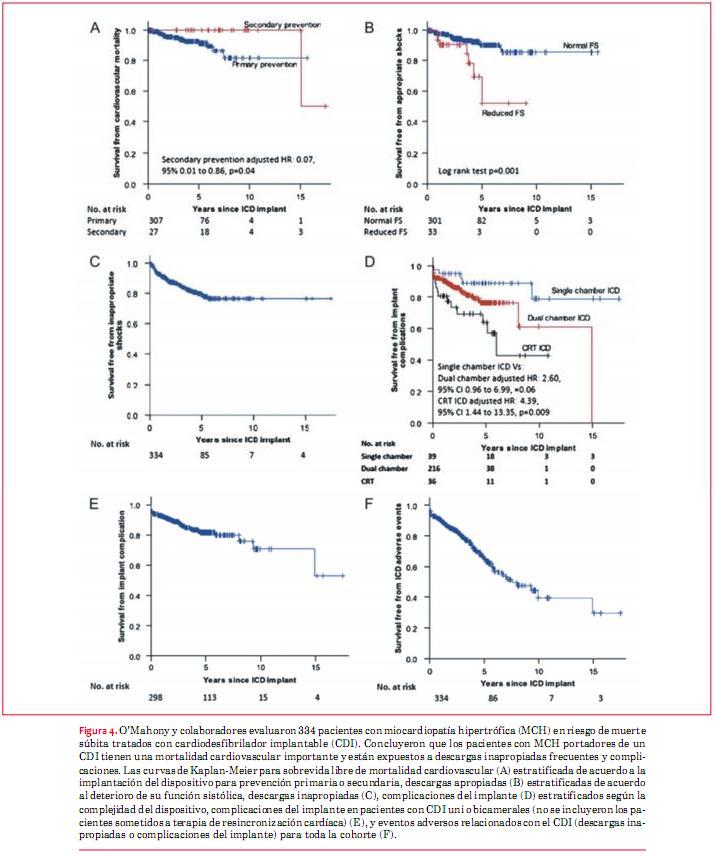
Una reciente revisión sistemática y metaanálisis de 27 estudios reportó una tasa de intervención apropiada del CDI de 3,3% por año con una tasa de descargas inapropiadas de 4,8% por año(29), pero en un estudio de un único centro de 334 pacientes con MCH, los pacientes aún tenían una mortalidad cardiovascular significativa (predominantemente por insuficiencia cardíaca) y presentaron descargas inapropiadas frecuentes y complicaciones del implante. Estos hallazgos sugieren que se necesitan nuevas estrategias para mejorar la selección de pacientes para implante de CDI y evitar el avance de la enfermedad en aquellos a los que se les coloca un dispositivo (figura 4)(30).
Actualmente la estimación del riesgo se basa en un puñado de marcadores de riesgo clínico de fácil obtención, utilizados para predecir la MSC y para guiar el implante de CDI(31,32). Pese a ello hay evidencia reciente que sugiere que la eficacia de este enfoque para distinguir a los individuos de alto y bajo riesgo es limitada(33). Además, recientemente se han propuesto varios factores pronósticos (tales como OTSVI y edad) que no fueron incluidos en dicha valoración(10,32-34). Hay datos recientes que también enfatizan la importancia de la edad en la estratificación del riesgo. En un estudio de 428 pacientes mayores de 60 años, 3,7% murieron debido a causas vinculadas a su MCH, como ACV embólico, insuficiencia cardíaca y trasplante. Se observaron eventos de MSC en cinco pacientes (1,2% o 0,2% por año). Los autores concluyen que los pacientes con MCH que sobreviven más allá de los 60 años tienen un menor riesgo de mortalidad relacionada con MCH y muerte súbita(35).
Los modelos económicos que analizan el impacto de las pruebas genéticas en la evaluación de familias con MCH concluyen que dichas pruebas tienen una buena relación costo-efectividad cuando se las combina con el screening clínico convencional(38,39). Estos modelos asumen que los algoritmos de riesgo provenientes de poblaciones de alto riesgo se pueden aplicar a poblaciones de bajo riesgo detectadas mediante el tamizaje y que el tratamiento preventivo con CDI es efectivo. Asimismo, estos modelos y muchos programas de screening clínico suponen una penetrancia relativamente alta de la enfermedad.
En un estudio que compara el screening clínico y las pruebas genéticas predictivas en niños y adolescentes, 90 pacientes con MCH y 361 familiares fueron seguidos durante 12 años(40). De un grupo de 12 jóvenes portadores de la mutación sin hipertrofia del VI, en la valoración inicial solo dos presentaron MCH durante el período del estudio, sugiriendo una penetrancia inesperadamente baja durante la adolescencia, período convencionalmente asociado con las mayores tasas de conversión fenotípica. Es importante destacar que los dos casos fueron diagnosticados a las edades de 26 y 28 años, lo que subraya la importancia del screening más allá de la adolescencia. Si estudios de mayor tamaño mostraran hallazgos similares, habría que reevaluar las estrategias convencionales de tamizaje clínico y el papel clínico de las pruebas genéticas, incluyendo su relación costo-efectividad.
La evidencia preliminar sugiere que el tamizaje clínico y las pruebas genéticas en niños y adultos no se asocian a consecuencias psicológicas adversas mayores(40). Habría que evaluar su efecto en los aspectos sociales y profesionales.
Relaciones entre genotipo y fenotipo
La definición de las relaciones clínicamente útiles entre el genotipo y el fenotipo sigue siendo elusiva en la MCH. Una revisión sistemática reciente comunicó una mayor prevalencia de antecedentes familiares de MCH y MSC, menor edad en el momento de la presentación, y un mayor espesor máximo del VI en individuos con una mutación en un gen sarcomérico, pero no se reportó ninguna diferencia en cuanto a las características clínicas al comparar las mutaciones MYBPC3 y MYH7. Sin embargo, estos datos estuvieron limitados por la inconsistencia del diseño de los estudios y el pequeño tamaño de muchas de sus cohortes(41).
Además de los estudios tradicionales acerca de la expresión clínica, varios grupos están utilizando miocitos cardíacos derivados de células madres pluripotentes inducidas (iPSC) humanas para estudiar la patogenia de la enfermedad. Se utilizaron técnicas de inmunotinción y patch clamping para identificar los fenotipos específicos de la enfermedad y diferencias en la toxicidad medicamentosa cardíaca entre las diferentes líneas celulares(42). El mismo grupo usó una técnica similar para demostrar que la restauración de la homeostasis del calcio impedía la aparición de hipertrofia de los miocitos y anomalías electrofisiológicas en miocardiocitos derivados de células madres pluripotentes portadoras de una mutación MYH7(43).
Miocardiopatía arritmogénica del ventrículo derecho
La miocardiopatía arritmogénica del ventrículo derecho (MAVD) se caracteriza clínicamente por presencia de arritmias, MSC e insuficiencia cardíaca progresiva. La pérdida de miocardiocitos y su sustitución por tejido fibroso o fibroadiposo constituyen marcadores histológicos de la enfermedad. La MAVD es provocada por mutaciones de los genes que codifican constituyentes del disco intercalado de miocardiocitos en una gran proporción de pacientes(44). El diagnóstico requiere la integración de datos de miembros de la familia, pruebas genéticas, electrocardiografía y técnicas imagenológicas(45). La MSC y el tratamiento de arritmias sintomáticas y de la insuficiencia cardíaca constituyen los principales retos.
Diagnóstico clínico de MAVD
Si bien la evidencia sugiere que la reciente modificación propuesta de los criterios diagnósticos ha mejorado la sensibilidad y especificidad diagnóstica(46,47), aún preocupa que puedan resultar demasiado sensibles en determinadas situaciones, fundamentalmente para atletas y en individuos de origen étnico negro africano, ya que muchos cambios estructurales y electrocardiográficos considerados normales en estos grupos también son criterios diagnósticos menores para MAVD(48,49).
Entre los métodos novedosos disponibles para detectar la expresión fenotípica temprana de la MAVD se han incluido enfoques inmunohistoquímicos y electrofisiológicos. Un reporte sugirió que la demostración inmunohistoquímica de una reducción de la señal de placoglobina en las biopsias miocárdicas tiene una sensibilidad de 85% y una especificidad de 57% para MAVD. Los autores sugirieron que se podría utilizar la prueba en el diagnóstico(50), pero no se ha estudiado su desempeño en los casos prefenotípicos, donde sería más valioso. Otro grupo de investigadores observó una marcada reducción de la señal inmunorreactiva para placoglobina en las uniones de los miocitos cardíacos en pacientes con sarcoidosis y miocarditis a células gigantes(51). Esto sugiere nuevos mecanismos patológicos en la miocarditis granulomatosa involucrando las proteínas desmosómicas y la intervención de citoquinas en la dislocación de la placoglobina de los desmosomas y en la aparición de arritmias en la MAVD.
Etiología
La MAVD se hereda como un rasgo autosómico dominante en hasta 50% de los casos(53) y es característica su penetrancia incompleta (incluyendo la penetrancia dependiente de la edad) y una expresión clínica variable. En el último año se ha subrayado la heterogeneidad genética de la MAVD por reportes de mutaciones nuevas en los genes para fosfolambán, desmocolina-2, TMEM43, CTNNA3 (a T catenina) y una delección de un gen en placofilina-2(54-60). Además, se han reportado mutaciones en genes hasta ahora asociadas con otras miocardiopatías en estudios de familias e individuos con MAVD. Estos incluyen la proteína no desmosómica lámina A/C(61). El papel de otros mecanismos genéticos y epigenéticos acerca de la expresión de la enfermedad sigue siendo un área de investigación activa(62).
Los avances en genética podrían mejorar la especificidad de los algoritmos diagnósticos en el futuro, pero existen numerosos retos al interpretar datos de las secuencias génicas. En un estudio de 427 controles y 93 casos índices de MAVD(63), se secuenciaron exones y sitios aceptores y donantes de empalmes en PKP2, DSR DSG2, DSC2 y TMEM43. Se identificaron mutaciones patogénicas probables en 58% de los casos de MAVD, pero también se las encontró en 16% de los controles. La mayoría (43%) de las mutaciones candidatas en los casos eran radicales (a saber: sitio de empalme, sin sentido, insertos y delecciones en el marco de lectura y corrimiento del marco de lectura) comparado con solo 0,5% de los controles, pero la frecuencia de las mutaciones de cambio de sentido fue similar en los casos (21%) y los controles (16%). Otros hallazgos importantes fueron una mayor frecuencia de variantes candidatas, en particular mutaciones de cambio de sentido, en controles caucásicos y no caucásicos (19,44% vs 5,83%) y números similares de variantes en los genes DSR DSC2 y TMEM43 en los grupos MAVD y control. Estos hallazgos ilustran el enfoque conservador que se debe aplicar al interpretar las variantes genéticas en MAVD.
El uso de iPSC como modelo de MAVD ha sido descrito recientemente(64). Los miocardiocitos de iPSC con mutación heterocigota de la placofilina-2 demostraron lipogénesis y apoptosis exageradas y déficits en el manejo del calcio en mutaciones homocigotas. Es posible que una mayor comprensión de estos fenómenos pueda llevar a desarrollar estrategias terapéuticas novedosas para la modificación de la enfermedad en el futuro.
Estrategias de manejo
Una vez que se diagnostica MAVD, el manejo debe incluir una valoración del riesgo de MSC, indicaciones de terapia medicamentosa y cambios del estilo de vida. Si bien los antiarrítmicos, como amiodarona y sotalol, se suelen indicar para reducir la carga arrítmica(44), hay poca evidencia de que mejoren la sobrevida o que alteren la historia natural de la enfermedad. Lo mismo se aplica al tratamiento de la disfunción sistólica del VI con inhibidores de la ECA y betabloqueantes.
Está descrito que el ejercicio físico y el deporte competitivo aumentan el riesgo de muerte súbita (65,66), por lo que no se recomienda su práctica(67,68). Más recientemente, el ejercicio ha sido asociado con una mayor penetrancia de la enfermedad y riesgo arrítmico en individuos con una mutación desmosómica. En 56 atletas de resistencia que presentaban la mutación se vio que era más probable que los criterios diagnósticos se cumplieran en el seguimiento y que los síntomas aparecieran a una edad más temprana que en portadores de la mutación sedentarios. Estos atletas también tuvieron una reducción de la sobrevida libre de taquicardia ventricular (TV), fibrilación ventricular e insuficiencia cardíaca(69). Estos hallazgos concuerdan con lo observado en el modelo murino de MAVD con defectos del gen de placofilina(70).
La ablación por catéter para tratar la TV recurrente en la MAVD se ha acompañado de altas tasas de recurrencia(71,72). Un estudio multicéntrico reciente enfocado en nuevas estrategias de ablación evaluó la recurrencia de TV luego de ablación por radiofrecuencia y su efecto sobre la carga de TV. Los autores comunican una importante reducción de la carga de TV y una supervivencia libre de TV más prolongada luego de la ablación epicárdica en comparación con un procedimiento endocárdico. Sin embargo, las tasas de recurrencia se mantienen considerablemente altas, con una ausencia de TV de 47% a un año(73). Estos datos sugieren que la ablación por catéter puede ayudar a un subgrupo de pacientes con TV incesante o frecuente refractaria a la terapia médica. Algunos datos sugieren que la TV atribuible a enfermedad localizada sería otra posible indicación(74).
Las actuales guías de AHA/ACC/ESC para el manejo de pacientes con arritmias ventriculares y la prevención de MSC recomiendan el implante de CDI en pacientes con MAVD en los que se haya documentado TV sostenida o fibrilación ventricular y que estén recibiendo tratamiento médico óptimo(75). Una revisión reciente de la literatura que investigó los resultados y las complicaciones de la implantación de CDI en MAVD incluyó a 610 pacientes. Durante el seguimiento de 3,8 años, los autores informan intervenciones apropiadas del CDI a una tasa de 9,5% por año, intervenciones inapropiadas 3,7%, y complicaciones (incluyendo mal funcionamiento o desplazamiento de los electrodos e infección) 20,3%(76). Nuevamente esto subraya la necesidad de realizar una correcta estratificación de riesgo para reducir a un mínimo la morbilidad secundaria a complicaciones relacionadas con el CDI. Es importante destacar que a los pacientes analizados en esta revisión se les colocó un CDI para prevención primaria o secundaria. Esto podría explicar, por lo menos parcialmente, la mayor tasa de intervenciones apropiadas.
Se estudió el creciente valor y el rol de la RMC en la estratificación del riesgo en 69 pacientes con mutaciones asociadas a MAVD (83% con mutaciones PKP-2) sin TV sostenida previa(77). Se encontraron anomalías eléctricas en 61% de los pacientes, 48% de los cuales tuvo una RMC anormal (definida como la presencia de por lo menos un criterio diagnóstico menor). Solo un paciente (4%) sin anomalías eléctricas tuvo un corazón anormal en los estudios imagenológicos iniciales. En un período de 5,8 ± 4,4 años únicamente se registraron episodios de TV sostenida en pacientes con anomalías en el ECG y la RMC. Los autores concluyeron que entre los portadores de mutaciones la presencia de anomalías, tanto eléctricas como en la RMC, identificaban pacientes de alto riesgo. Un estudio similar, evaluando el pronóstico en 369 pacientes que cumplieron con por lo menos un criterio diagnóstico menor o mayor para MAVD, describe el valor predictivo positivo de la RMC anormal. El valor predictivo negativo de una RMC normal fue de 98,8% para un período de seguimiento de 4,3 ± 1,5 años(78).
La búsqueda de biomarcadores que permitan un diagnóstico temprano y una estratificación del riesgo es un área activa de investigación. Por ejemplo, las bajas concentraciones séricas del bridging integrator 1 (BIN-1), una proteína asociada a la membrana, se vincularon a presencia de arritmia ventricular y reducción del estado funcional en una pequeña cohorte de 24 pacientes con MAVD(79).
Una novedosa estrategia de predicción de riesgo en portadores de mutaciones desmosómicas asociadas a MAVD propone utilizar la evaluación del árbol genealógico y la información proveniente del ECG y el Holter(80). Para estratificar el riesgo de TV sostenida se utilizaron las características fenotípicas. Los investigadores incluyeron 215 pacientes con una media de seguimiento de siete años. Se estratificó el riesgo de los pacientes según la presencia de anomalías de repolarización y despolarización en el ECG. La sobrevida libre de eventos a los cinco años fue de 33% en el grupo de alto riesgo contra 97% en el grupo de bajo riesgo.
Miocardiopatía dilatada
La miocardiopatía dilatada (MCD) es una de las patologías del músculo cardíaco más frecuentes en los países desarrollados. Se la define por la presencia de disfunción sistólica y dilatación del VI en ausencia de infarto de miocardio previo. En los últimos años han cobrado mucha importancia los estudios que destacan la importancia de la genética en la etiología de las formas hereditarias y las aparentemente adquiridas de MCD. Los tratamientos sintomáticos y con impacto pronóstico estándar de la insuficiencia cardíaca constituyen el principal pilar del manejo de los pacientes, pero en los últimos tiempos se le ha prestado más atención a la importancia de la etiología para guiar el manejo.
Subtipos genéticos de MCD
Varios estudios han examinado la historia natural de la MCD provocada por mutaciones en el gen de la lámina A/C (LMNA). Se acompaña de alteraciones de la conducción, arritmias auriculares, insuficiencia cardíaca y muerte súbita, y se debe sospechar su existencia cuando la MCD se acompaña de elevación de la creatininquinasa sérica, alteraciones de la conducción o arritmias frecuentes. En estos pacientes, la evidencia sugiere que el implante de CDI debería ser considerado con menor umbral que en otros casos de MCD(81-83). Una cohorte multicéntrica de 269 pacientes con la mutación LMNA identificó como factores de riesgo para arritmias ventriculares malignas la presencia de TV no sostenida, fracción de eyección del ventrículo izquierdo (FEVI) <45%, sexo masculino y mutaciones que no sean de cambio de sentido (non-missense)(84). Algunas autoridades sugieren que habría que considerar el implante de CDI aun ante una expresión cardíaca leve.
La importancia de los factores epigenéticos (a saber, los procesos que alteran la activación de genes sin cambiar la secuencia del ADN) también ha sido destacada desde 2011. En un estudio que compara la metilación del ADN cardíaco de todo el genoma en pacientes con MCD idiopática y controles, Haas y colaboradores detectaron diferencias en la metilación de los genes implicados en los mecanismos de insuficiencia cardíaca. Se acompañaron de diferencias en la expresión del ARNm, un hallazgo fortalecido más aun por estudios en el pez cebra(90). La investigación epigenética está particularmente avanzada en el estudio del cáncer, y aportaría posibles biomarcadores diagnósticos y blancos terapéuticos(91-93). Todavía no se conoce su potencial en la genética cardiovascular.
La predisposición genética al daño miocárdico inflamatorio también está surgiendo como un tema importante(94). El virus Coxsackie, una causa común de miocarditis, provoca la proteólisis de la distrofina en los miocardiocitos infectados(95-97). Los defectos genéticos del complejo distrofina-glicoproteína, asociados con la distrofia muscular, frecuentemente provocan una MCD cuyas manifestaciones en la RMC a menudo son indistinguibles de la miocarditis(98). En otro estudio, la presencia de variantes en los receptores tipo Toll, que intervienen en la respuesta inmunitaria innata, se acompañaron de una peor función cardíaca en 158 pacientes(99). Finalmente, Meder y colaboradores(100) presentan datos que asocian el locus que contiene genes de histocompatibilidad mayor (MHC I y II) con la MCD. Los autores identificaron múltiples polimorfismos de un único nucleótido en el cromosoma 6p21. Se identificó un locus específico y se halló una asociación con genes ubicados cerca que codificaban para receptores de cadena pesada del complejo de histocompatibilidad mayor clase I y clase II.
Predicción de los resultados en la MCD
Al igual que con otras miocardiopatías, el papel de la RMC para predecir los resultados es un área activa de estudio en MCD. Las últimas evidencias sugieren que la presencia de RTG intramiocárdica identifica una cohorte de MCD con un mayor riesgo de mortalidad. Durante un período de seguimiento con una mediana de un poco más de cinco años, murió el 27% de los 142 pacientes con MCD y RTG comparado con 11% de los 330 pacientes con MCD sin RTG(101). No queda claro cuál es la base fisiopatológica de esta diferencia.
En un futuro cercano se espera disponer de datos que describan la utilidad diagnóstica y pronóstica de la RMC para otras anomalías tisulares como edema, fibrosis difusa o desarreglo de los miocitos.
Algunos de los datos más importantes surgidos desde 2011 provienen de niños con MCD. En una población de un registro pediátrico de 1.803 pacientes, se describe una tasa de trasplante cardíaco a cinco años de 29%, 12,1% para muerte cardíaca no súbita y 2,4% para MSC. El modelo de estratificación de riesgo de MSC basado en los datos ecocardiográficos tiene una sensibilidad de 86% y especificidad de 57%. Entre los factores de importancia se incluyen la dilatación del VI, la edad en el momento del diagnóstico y el adelgazamiento de la pared posterior(102).
Otro estudio que incluyó a 175 pacientes pediátricos con MCD informa muerte o trasplante en 26% de los casos a un año del diagnóstico, con una tasa de sobrevida libre de muerte o trasplante de 56% veinte años después del diagnóstico. El aumento del riesgo de muerte o trasplante se asoció a la edad al momento del diagnóstico, presencia de miocardiopatía familiar y una menor fracción de acortamiento del VI a nivel basal(103).
La terapia con células madre ha sido un tema importante en el curso de los últimos años. Un estudio controlado aleatorizado de cinco años de seguimiento de 110 pacientes con MCD muestra una mejor función del VI, mejor tolerancia al ejercicio y una mayor sobrevida a largo plazo en pacientes sometidos a trasplante intracoronario de células madre. La mortalidad total fue de 14% en el grupo de células madre contra 35% en los controles, con tasas de falla de la bomba de 5% contra 18%. No hubo diferencia en la tasa de muerte súbita(104). Una revisión sistemática de 29 estudios preclínicos y 15 clínicos estudiaron la terapia con células madre como un tratamiento para MCD. En el curso del seguimiento la mayoría de los estudios mostró una mejora modesta de la FEVI después de la terapia celular. Dada la gran heterogeneidad de los criterios de inclusión, procedimientos y medidas de resultados, se enfatizó en la necesidad de ensayos controlados aleatorizados(105).
El efecto del ejercicio a corto plazo ha sido evaluado en pacientes con MCD luego de un período de ocho semanas de ejercicio. Se comunicó una importante mejora de la función cardíaca en reposo y luego del ejercicio, siendo los pacientes sedentarios los que tenían la mayor mejora(106).
La importancia de la activación inmunitaria es otro punto al que dirigir la terapia en casos de MCD. La atorvastatina a bajas dosis redujo los niveles de citoquinas inflamatorias (IL-6, TNFa), ácido úrico y NT-pro BNP en una pequeña cohorte de pacientes con MCD(107).
La evidencia general sugiere una mejora en la supervivencia de los pacientes con MCD idiopática. Un estudio de 603 pacientes en el curso de tres décadas arroja evidencia acerca del impacto de las guías de práctica clínica sobre la morbilidad y la mortalidad. Los pacientes se subdividieron en cuatro períodos de registro y se comunicó una reducción del riesgo de 42% por intervalo de registro con respecto a la mortalidad relacionada con la insuficiencia cardíaca y la muerte súbita(108).
Resumen
Las miocardiopatías siguen siendo un área de intenso interés en la literatura. Aunque el espectro de las enfermedades es considerable y sigue ampliándose, los aspectos a considerar son muy parecidos entre los subtipos de miocardiopatías, enfatizándose más el diagnóstico preciso, la estratificación de la enfermedad y la terapia según la etiología. Los avances en estas áreas dependerán de descubrimientos científicos y la aplicación de tecnologías ómicas, pero es probable que los mayores esclarecimientos surjan de colaboraciones multicéntricas de gran escala. Todo sugiere que en los próximos años se harán avances considerables.
Colaboración: todos los autores contribuyeron en lo siguiente: concepción y diseño, adquisición e interpretación de los datos; redacción del artículo o revisión crítica del contenido intelectual importante y aprobación final de la versión a publicar.
Conflicto de intereses: OPG recibió apoyo para investigación de la British Heart Foundation. Ningún otro autor manifestó tener conflictos de interés.
Bibliografía
1. Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995;92:785-9.
2. Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 2004;363:1881-91.
3. Guttmann OP, Rahman MS, O’Mahony C, et al. Atrial ?brillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014;100:6 465-72.
4. Puntmann VO, Voigt T, Chen Z, et al. Native T1 mapping in differentiation of normal myocardium from diffuse disease in hypertrophic and dilated cardiomyopathy. JACC Cardiovasc Imaging 2013;6:475-84.
5. Sado DM, Flett AS, Banypersad SM, et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart 2012;98:1436-41.
6. Sado DM,
7. Ferreira VM,
8. Valente AM, Lakdawala NK, Powell AJ, et al. Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet 2013;6:230-7.
10. Greulich S, Schumm J, Grun S, et al. Incremental value of late gadolinium enhancement for management of patients with hypertrophic cardiomyopathy. Am J Cardiol 2012;110:1207-12.
11. Green JJ, Berger JS, Kramer CM, et al. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging 2012;5:370-7.
12. van Veldhuisen DJ, Linssen GC, Jaarsma T, et al. B-type natriuretic peptide and prognosis in heart failure patients with preserved and reduced ejection fraction. J Am Coll Cardiol 2013;61:1498-506.
13. Geske JB, McKie PM, Ommen SR, et al. B-type natriuretic Peptide and survival in hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;61:2456-60.
14. D’Amato R, Tomberli B, Castelli G, et al. Prognostic value of N-terminal pro-brain natriuretic peptide in outpatients with hypertrophic cardiomyopathy. Am J Cardiol 2013;112:1190-6.
15. Coats CJ, Gallagher MJ, Foley M, et al. Relation between serum N-terminal pro-brain natriuretic peptide and prognosis in patients with hypertrophic cardiomyopathy. Eur Heart J 2013;34:2529-37.
16. Kubo T, Kitaoka H, Yamanaka S, et al. Signi?cance of high-sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;62:1252-9.
17. Nistri S, Olivotto I, Maron MS, et al. Beta blockers for prevention of exercise-induced left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. Am J Cardiol 2012;110: 715-9.
18. Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to ?rst-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013;6:694-702.
19. Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012;126: 2374-80.
20. Jensen MK, Prinz C, Horstkotte D, et al. Alcohol septal ablation in patients with hypertrophic obstructive cardiomyopathy: low incidence of sudden cardiac death and reduced risk pro?le. Heart 2013;99:1012-7.
21. Orme NM, Sorajja P, Dearani JA, et al. Comparison of surgical septal myectomy to medical therapy alone in patients with hypertrophic cardiomyopathy and syncope. Am J Cardiol 2013;111:388-92.
22. Iacovoni A, Spirito P, Simon C, et al. A contemporary European experience with surgical septal myectomy in hypertrophic cardiomyopathy. Eur Heart J 2012;33:2080-7.
23. Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular out?ow tract obstruction. Circulation 2013;128:209-16.
24. Galve E, Sambola A, Saldana G, et al. Late bene?ts of dual-chamber pacing in obstructive hypertrophic cardiomyopathy: a 10-year follow-up study. Heart 2010;96:352-6.
25. Mohiddin SA, Page SP. Long-term bene?ts of pacing in obstructive hypertrophic cardiomyopathy. Heart 2010;96:328-30.
26. Kappenberger LJ, Linde C, Jeanrenaud X, et al. Clinical progress after randomized on/off pacemaker treatment for hypertrophic obstructive cardiomyopathy. Pacing in Cardiomyopathy (PIC) Study Group. Europace 1999;1:77-84.
27. Maron BJ, Nishimura RA, McKenna WJ, et al. Assessment of permanent dual-chamber pacing as a treatment for drug-refractory symptomatic patients with obstructive hypertrophic cardiomyopathy. A randomized, double-blind, crossover study (M-PATHY). Circulation 1999;99:2927-33.
28. Qintar M, Morad A, Alhawasli H, et al. Pacing for drug-refractory or drug-intolerant hypertrophic cardiomyopathy. Cochrane Database Syst Rev 2012; (5):CD008523.
29. Schinkel AF, Vriesendorp PA, Sijbrands EJ, et al. Outcome and complications after implantable cardioverter de?brillator therapy in hypertrophic cardiomyopathy: systematic review and meta-analysis. Circ Heart Fail 2012;5:552–9.
30. O’Mahony C, Lambiase PD, Quarta G, et al. The long-term survival and the risks and bene?ts of implantable cardioverter de?brillators in patients with hypertrophic cardiomyopathy. Heart 2012;98: 116-25.
31. Bos JM, Maron BJ, Ackerman MJ, et al. Role of family history of sudden death in risk strati?cation and prevention of sudden death with implantable de?brillators in hypertrophic cardiomyopathy. Am J Cardiol 2010;106:1481-6.
32. Christiaans I, van Engelen K, van Langen IM, et al. Risk strati?cation for sudden cardiac death in hypertrophic cardiomyopathy: systematic review of clinical risk markers. Europace 2010;12:313-21.
33. O’Mahony C, Tome-Esteban M, Lambiase PD, et al. A validation study of the 2003 American College of Cardiology/European Society of Cardiology and 2011 American College of Cardiology Foundation/American Heart Association risk strati?cation and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart 2013;99:534-41.
35. Maron BJ, Rowin EJ, Casey SA, et al. Risk strati?cation and outcome of patients with hypertrophic cardiomyopathy >=60 years of age. Circulation 2013;127:585-93.
36. Moak JP, Leifer ES, Tripodi D, et al. Long-term follow-up of children and adolescents diagnosed with hypertrophic cardiomyopathy: risk factors for adverse arrhythmic events. Pediatr Cardiol 2011;32: 1096-105.
37. Maron BJ, Spirito P, Ackerman MJ, et al. Prevention of sudden cardiac death with implantable cardioverter-de?brillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;61:1527-35.
38. Ingles J, McGaughran J, Scuffham PA, et al. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012;98:625–30.
39. Wordsworth S, Leal J, Blair E, et al. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 2010;31:926-35.
40. Jensen MK, Havndrup O, Christiansen M, et al. Penetrance of hypertrophic cardiomyopathy in children and adolescents: a 12-year follow-up study of clinical screening and predictive genetic testing. Circulation 2013;127:48-54.
41. Lopes LR, Rahman MS, Elliott PM. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013;99:1800-11.
42. Liang P, Lan F, Lee AS, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-speci?c patterns of cardiotoxicity. Circulation 2013; 127:1677-91.
43. Lan F, Lee AS, Liang P, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-speci?c induced pluripotent stem cells. Cell Stem Cell 2013;12:101-13.
44. Corrado D, Basso C, Pilichou K, et al. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/ dysplasia. Heart 2011;97:530-9.
45. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modi?cation of the Task Force Criteria. Eur Heart J 2010;31:806-14.
46. Protonotarios N, Anastasakis A, Antoniades L, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur Heart J 2011;32:1097-104.
47. Quarta G, Muir A, Pantazis A, et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 2011;123:2701–9.
48. Basso C, Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy in athletes: diagnosis, management, and recommendations for sport activity. Cardiol Clin 2007;25:415-22, vi.
49. Zaidi A, Ghani S, Sheikh N, et al. Clinical signi?cance of electrocardiographic right ventricular hypertrophy in athletes: comparison with arrhythmogenic right 762 Guttmann OP, et al. Heart 2014;100:756–764.
50. Munkholm J, Christensen AH, Svendsen JH, et al. Usefulness of immunostaining for plakoglobin as a diagnostic marker of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2012;109:272-5.
51. Asimaki A, Tandri H, Duffy ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol 2011;4:743-52.
52. Gomes J, Finlay M, Ahmed AK, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J 2012;33:1942-53.
53. Cox MG, van der Zwaag PA, van der Werf C, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011;123:2690-700.
54. Li Mura IE, Bauce B, Nava A, et al. Identi?cation of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. Eur J Hum Genet 2013;21:1226-31.
55. van der Zwaag PA, van Rijsingen IA, de Ruiter R, et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J 2013;21:286-93.
56. Baskin B, Skinner JR, Sanatani S, et al. TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum Genet 2013;132: 1245-52.
58. Gerull B, Kirchner F, Chong J, et al. A homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet 2013;6:327–36.
59. Haywood AF,
60. Groeneweg JA, van der Zwaag PA, Jongbloed JD, et al. Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Heart Rhythm 2013; 10:548-59.
61. Quarta G, Syrris P, Ashworth M, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2012; 33:1128-36.
62. Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol 2013;61:1945-8.
63. Kapplinger JD, Landstrom AP,
64. Kim C, Wong J, Wen J, et al. Studying arrhythmogenic right ventricular dysplasia with patient-speci?c iPSCs. Nature 2013;494:105-10.
65. Corrado D, Basso C, Rizzoli G, et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol 2003;42:1959-63.
66. Fabritz L, Fortmuller L, Yu TY, et al. Can preload-reducing therapy prevent disease progression in arrhythmogenic right ventricular cardiomyopathy? Experimental evidence and concept for a clinical trial. Prog Biophys Mol Biol 2012;110:340-6.
67. Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109:2807-16.
68. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol 2005;45:1340-5.
69. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290-7.
70. Fabritz L, Hoogendijk MG, Scicluna BP, et al. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J Am Coll Cardiol 2011;57:740-50.
71. Dalal D, Jain R, Tandri H, et al. Long-term ef?cacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/ cardiomyopathy. J Am Coll Cardiol 2007;50:432-40.
72. Verma A, Kilicaslan F, Schweikert RA, et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation 2005;111:3209-16.
73. Philips B, Madhavan S, James C, et al. Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/ cardiomyopathy. Circ Arrhythm Electrophysiol 2012;5:499-505.
74. Basso C, Corrado D, Bauce B, et al. Arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol 2012;5:1233-46.
75. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol 2006;48:e247-346.
76. Schinkel AF. Implantable cardioverter de?brillators in arrhythmogenic right ventricular dysplasia/cardiomyopathy: patient outcomes, incidence of appropriate and inappropriate interventions, and complications. Circ Arrhythm Electrophysiol 2013;6:562-8.
77. Te Riele AS, Bhonsale A, James CA, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk strati?cation of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1761-9.
78. Deac M, Alpendurada F, Fanaie F, et al. Prognostic value of cardiovascular magnetic resonance in patients with suspected arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol 2013;168: 3514-21.
80. Bhonsale A, James CA, Tichnell C, et al. Risk strati?cation in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol 2013;6:569-78.
81. van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl) 2005;83:79-83.
82. Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med 2006;354: 209-10.
83. Taylor MR, Fain PR, Sinagra G, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003;41:771–80.
84. van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol 2012;59(5):493-500.
85. Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619-28.
86. Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle ?lament titin, cause familial dilated cardiomyopathy. Nat Genet 2002;30:201-4.
87. Gerull B, Atherton J, Geupel A, et al. Identi?cation of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med (Berl) 2006;84:478-83.
88. Siu BL, Niimura H, Osborne JA, et al. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation 1999;99:1022-6.
89. Satoh M, Takahashi M, Sakamoto T, et al. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 1999;262:411-17.
90. Haas J,
91. Coppede F. Epigenetic biomarkers of colorectal cancer: focus on DNA methylation. Cancer Lett 2014;342:238-47.
92. Litzow MR. Novel therapeutic approaches for acute lymphoblastic leukemia. Hematol Oncol Clin North Am 2011;25:1303-17.
93. Herceg Z, Vaissiere T. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics 2011;6:804-19.
94. Mavrogeni S, Spargias C, Bratis C, et al. Myocarditis as a precipitating factor for heart failure: evaluation and 1-year follow-up using cardiovascular magnetic resonance and endomyocardial biopsy. Eur J Heart Fail 2011;13: 830-7.
95. Badorff C, Knowlton KU. Dystrophin disruption in enterovirus-induced myocarditis and dilated cardiomyopathy: from bench to bedside. Med Microbiol Immunol 2004;193:121-6.
96. Badorff C, Fichtlscherer B, Rhoads RE, et al. Nitric oxide inhibits dystrophin proteolysis by coxsackieviral protease 2A through S-nitrosylation: a protective mechanism against enteroviral cardiomyopathy. Circulation 2000;102: 2276-81.
97. Badorff C, Lee GH, Knowlton KU. Enteroviral cardiomyopathy: bad news for the dystrophin-glycoprotein complex. Herz 2000;25:227-32.
98. Mavrogeni S, Papavasiliou A, Spargias K, et al. Myocardial inflammation in Duchenne Muscular Dystrophy as a precipitating factor for heart failure: a prospective study. BMC Neurol 2010;10:33.
99. Riad A, Meyer zu SH, Weitmann K, et al. Variants of Toll-like receptor 4 predict cardiac recovery in patients with dilated cardiomyopathy. J Biol Chem 2012;287:27236-43.
100. Meder B, Ruhle F, Weis T, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J 2013.
101. Gulati A, Jabbour A, Ismail TF, et al. Association of ?brosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013;309:896-908.
102. Pahl E, Sleeper LA, Canter CE, et al. Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol 2012;59:607-15.
103. Alexander PM,
104. Vrtovec B, Poglajen G, Lezaic L, et al. Effects of intracoronary CD34+ stem cell transplantation in nonischemic dilated cardiomyopathy patients: 5-year follow-up. Circ Res 2013;112:165-73.
106. Holloway CJ, Dass S, Suttie JJ, et al. Exercise training in dilated cardiomyopathy improves rest and stress cardiac function without changes in cardiac high energy phosphate metabolism. Heart 2012;98:1083-90.
107. Bielecka-Dabrowa A, Mikhailidis DP, Rizzo M, et al. The in?uence of atorvastatin on parameters of in?ammation left ventricular function, hospitalizations and mortality in patients with dilated cardiomyopathy—5-year follow-up. Lipids Health Dis 2013; 12:47.
108. Castelli G, Fornaro A, Ciaccheri M, et al. Improving survival rates of patients with idiopathic dilated cardiomyopathy in


{kind=link}