










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkRevista Uruguaya de Cardiología
versão On-line ISSN 1688-0420
Rev.Urug.Cardiol. vol.28 no.3 Montevideo dez. 2013
Artículo de revisión
Antiagregación plaquetaria en los síndromes coronarios agudos
Dres. Gustavo Vignolo1, Rafael Mila2
1. Profesor Agregado de Cardiología.
2. Profesor Adjunto de Cardiología.
Palabras clave:
SíNDROME CORONARIO AGUDO
ANTIAGREGANTES PLAQUETARIOS
DOBLE ANTIAGREGACIóN PLAQUETARIA
Key words:
ACUTE CORONARY SYDROME
ANTIAGGREGANTS, PLATELET
DOUBLE ANTIAGGREGANTS PLATELET
Introducción
Las plaquetas juegan un rol central en el proceso trombótico que sigue a la ruptura, fisura o erosión de una placa aterosclerótica(1). El tratamiento antitrombótico es la piedra angular del tratamiento de los eventos aterotrombóticos, particularmente en los síndromes coronarios agudos (SCA). A partir del descubrimiento del mecanismo de acción de la aspirina en la actividad plaquetaria se ha producido un gran avance en el conocimiento de los mecanismos de activación e inhibición de las plaquetas.
La terapia antiplaquetaria dual con aspirina y clopidogrel se ha consolidado como el tratamiento estándar frente a un SCA tanto con sobreelevación persistente del segmento ST (SCAcST) como sin sobreelevación del ST (SCAsST). Sin embargo, un número considerable de pacientes presenta recurrencia de eventos bajo este régimen, lo que llevó al desarrollo de fármacos antiplaquetarios nuevos de mayor potencia, algunos recientemente aprobados y otros aún en desarrollo.
Analizaremos inicialmente la fisiopatología de la trombosis mediada por plaquetas y luego cada uno de los agentes antiplaquetarios de uso clínico relevante. En cada caso se expondrán las características farmacológicas y la evidencia más relevante, para finalmente resumir las recomendaciones respecto a su uso en los SCA.
1. Fisiopatología de la trombosis mediada por plaquetas
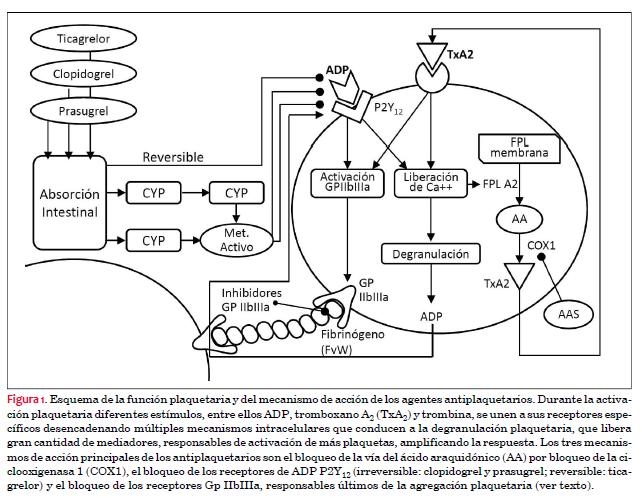
La aterotrombosis es un hecho central en la evolución y complicación de la enfermedad aterosclerótica. El evento central en la patogenia de los SCA es la disrupción de una lesión aterosclerótica con formación de un trombo endoluminal como consecuencia de la puesta en marcha de dos sistemas paralelos: la función plaquetaria y la cascada de la coagulación. El componente plaquetario del trombo es consecuencia de la adhesión, activación y agregación de las plaquetas(1). Nos referiremos de manera esquemática y resumida a estos tres fenómenos plaquetarios:
- Adhesión. La injuria del endotelio vascular por ruptura o erosión de placa expone el subendotelio, lo que permite al factor de von Willebrand (vWF) unirse al colágeno. El colágeno unido al vWF funciona como punto de anclaje para las plaquetas, lo que inicia la fase de adhesión plaquetaria(2).
- Activación. Como consecuencia de la adhesión plaquetaria se desencadena la activación plaquetaria. Este proceso resulta de la liberación autócrina y parácrina de mediadores, como el difosfato de adenosina (ADP), TXA2, y la producción local de trombina. El ADP se une a sus receptores de membrana específicos, P2Y1 y P2Y12, en la superficie plaquetaria. La activación del receptor P2Y1 conduce al aumento de la concentración intracelular de calcio, mientras que la activación del receptor P2Y12 lleva a una disminución en los niveles de monofosfato de adenosina cíclico (cAMP). El receptor P2Y12 juega un rol central en la amplificación y el mantenimiento de la respuesta al ADP y constituye el sitio de acción de los inhibidores P2Y12, es decir, clopidogrel, prasugrel y ticagrelor(3). El TXA2 es producido por las plaquetas activadas a partir del ácido araquidónico por la vía de la ciclooxigenasa, de gran importancia terapéutica dado que esta vía es bloqueada por el ácido acetilsalicílico (AAS). Luego de su unión a receptores de TXA2 (a o b), la cascada de eventos lleva al incremento en la concentración intracelular de calcio(4). Un tercer mediador plaquetario de importancia central es la trombina, sintetizada rápidamente desde la protrombina plasmática en los sitios de daño vascular. La unión de la trombina a los receptores de proteasa activada (PAR-1 y 4) resulta en cambios en la morfología de la plaqueta, incremento en la concentración intracelular de calcio y disminución de la concentración de cAMP. La trombina es además responsable de la generación de fibrina(5).
- Agregación. El efecto neto del incremento de la concentración intracelular de calcio y de la disminución del cAMP es la activación de la GP IIb/IIIa, mediador final de la agregación plaquetaria. Este fenómeno es mediado por la unión de la GP IIb/IIIa a sustratos solubles que incluyen al fibrinógeno y wWF. El fibrinógeno forma puentes entre las plaquetas activadas estabilizando de esta manera al trombo mediante el proceso de agregación plaquetaria. Los trombos ricos en plaquetas lucen “blancos” y habitualmente son suboclusivos, mientras que los trombos “rojos” (ricos en fibrina) se forman en el sector luminal del trombo blanco y se asocian típicamente a oclusión total del vaso(6).
- inhibidores de la ciclooxigenasa-1 (COX-1): AAS.
- inhibidores de los receptores P2Y12:
- tienopiridinas: ticlopidina, clopidogrel, prasugrel.
- no tienopiridina (ciclopentiltriazolopirimidina): ticagrelor.
- inhibidores de la glicoproteína IIb/IIIa (inhibidores GP IIb/IIIa): abciximab, eptifibatide y tirofiban(7-10).
2. Agentes antiplaquetarios en los síndromes coronarios agudos
Actualmente existen tres clases de fármacos antiplaquetarios aprobados para el uso clínico y recomendados por las guías de práctica clínica:
Analizaremos inicialmente en forma individual cada uno de los antiagregantes de relevancia clínica en los SCA y luego revisaremos las recomendaciones sobre su uso en este contexto clínico.
El mecanismo de acción de los diferentes fármacos antiplaquetarios se ilustra en la figura 1.
2.a. Ácido acetilsalicílico
El AAS bloquea de forma irreversible la enzima COX-1 mediante acetilación. Esta inhibición enzimática previene la conversión de ácido araquidónico en prostaglandina G2/H2 y la subsecuente síntesis de TXA2 mediante la acción de la tromboxano sintetasa. Las características farmacológicas más relevantes del AAS se resumen en la tabla 1.
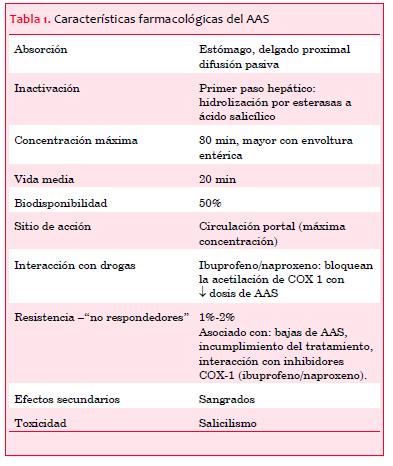
El AAS es el fármaco más simple y con mejor relación costo-efectividad de la cardiología, con una larga historia en el arsenal terapéutico médico aunque con una más breve trayectoria en medicina cardiovascular. Existe un amplio espectro de afecciones que se benefician del uso de AAS y que incluyen algunas situaciones de prevención primaria, todos los casos de prevención secundaria, todas las formas de revascularización coronaria y todos los tipos de SCA.
Múltiples dosis de AAS han sido estudiadas, comprobándose similar eficacia para un amplio rango de dosis, con mayor riesgo de sangrado para las más elevadas. Por esto, se recomienda la utilización de dosis en torno a 100 mg/día(11). Recientemente, el estudio CURRENT-OASIS 7 comparó la utilización de dosis de AAS “altas” (300-325 mg/día por un mes) versus “bajas” (75-100 mg/día) en pacientes con SCA sin y con sobreelevación del segmento ST. No se comprobaron diferencias significativas entre ambos regímenes en relación con muerte, infarto agudo de miocardio (IAM) o accidente cerebrovascular (ACV) a 30 días, aunque se observó un aumento de sangrados menores asociado a las dosis altas, incluyendo sangrado digestivo(12). Estos resultados se comprobaron tanto en la población general como en los pacientes tratados con angioplastia coronaria, por lo que parece razonable la recomendación de dosis de mantenimiento de AAS entre 75 y 100 mg/día. Esto es aplicable tanto a los SCAsST como a los SCAcST, ya que ambas poblaciones estuvieron representadas en el estudio CURRENT.
El uso de aspirina, incluso a dosis £ 325 mg, se asocia a un incremento de dos a cuatro veces en el riesgo de úlcera péptica complicada o sintomática. La presencia de Helicobacter pylori aumenta la toxicidad gastrointestinal y la erradicación del mismo reduce la incidencia de esta complicación(13). Una revisión sistemática de dosis diferentes de aspirina demostró un incremento del riesgo de sangrado con dosis crecientes, sin beneficio adicional, en caso de utilizar dosis superiores a 81 mg/día(14). El aumento del riesgo de sangrado no depende del agregado de clopidogrel; dosis altas de AAS en monoterapia se asocia a mayor riesgo de sangrado que a la combinación de bajas dosis de AAS sumado a clopidogrel(15).
A pesar del efecto beneficioso del AAS, algunos pacientes persisten aún en riesgo de eventos trombóticos debido a una inhibición insuficiente de las plaquetas, lo que se ha vinculado a resistencia a la aspirina. Si bien no hay acuerdo en cuanto a la definición de laboratorio de resistencia a la aspirina, un metaanálisis reportó una incidencia de resistencia a la aspirina de 27% con un incremento del riesgo de eventos cardiovasculares mayores a largo plazo en este grupo de pacientes(16). Cuando se analiza en profundidad el fenómeno de resistencia a la aspirina mediante técnicas de laboratorio específicas que aíslan la actividad de la COX, se encuentra que la resistencia es muy infrecuente cuando los pacientes adhieren correctamente a la medicación. Se acepta actualmente que la mayor parte de los casos de resistencia a la aspirina están relacionados principalmente con el incumplimiento terapéutico más que con una verdadera resistencia a la acción del fármaco(2). De hecho, la no adherencia a la medicación luego de un IAM ha demostrado asociarse directamente con mortalidad(17). Otros factores que pueden incrementar la probabilidad de resistencia a la acción de la aspirina son la coadministración de antiinflamatorios no esteroideos que pueden inhibir competitivamente el acceso de la aspirina a la acetilación de la COX-1, hiperplaquetosis, enfermedades inflamatorias crónicas como la artritis reumatoidea, diabetes mellitus y otras condiciones en que la existencia de un estado pro inflamatorio y pro trombótico sobrepasan los efectos de la aspirina sobre la COX-1(2).
2.b. Inhibidores de los receptores P2Y12
2.b.I. Tienopiridinas
Las tienopiridinas inhiben de forma selectiva e irreversible el receptor P2Y12 durante toda la vida de la plaqueta. Son pro drogas que requieren biotransformación hepática al correspondiente metabolito activo(18). El desarrollo de estos fármacos ha impactado favorablemente en la evolución del SCA. La ticlopidina fue la primera clínicamente disponible y a pesar de su eficacia, su uso fue limitado debido a la incidencia frecuente de eventos adversos potencialmente fatales (neutropenia y púrpura trombocitopénico trombótico) y al desarrollo del clopidogrel, una tienopiridina con un mejor perfil de seguridad. Recientemente ha sido introducido en el mercado el prasugrel, una tienopiridina de tercera generación, más potente, que constituye una nueva opción en el tratamiento de los SCA. Analizaremos sucesivamente clopidogrel y prasugrel.
2.b.I.a. Clopidogrel
El clopidogrel constituyó un gran avance en el tratamiento antiplaquetario y representó el tratamiento estándar del SCA en la última década hasta la reciente aprobación de los nuevos antiplaquetarios: prasugrel y ticagrelor.
El clopidogrel se absorbe a nivel intestinal y es metabolizado por dos vías competitivas en el hígado. En la primera, el clopidogrel es rápidamente metabolizado por la carboxilesterasa humana 1 a un metabolito ácido inactivo. Aproximadamente 75% del clopidogrel administrado se procesa mediante esta vía(19). En la segunda, el clopidogrel es metabolizado en el hígado por la citocromo P450 en un proceso de dos pasos. El primer paso transforma el clopidogrel en 2-oxo-clopidogrel (enzimas: CYP2C19, CYP1A2 y CYP2B6) y el segundo paso lo transforma en su metabolito activo (enzimas: CYP2B6, CYP3A, CYP2C9 y CYP2C19). Luego de su bioactivación, el metabolito activo se une de forma irreversible al receptor P2Y12. El 2-oxo-clopidogrel también puede ser transformado a su metabolito inactivo. Como sustrato de la CYP2C, el clopidogrel es susceptible a interacciones con otros fármacos(2). Las características farmacológicas más relevantes del clopidogrel se resumen en la tabla 2.
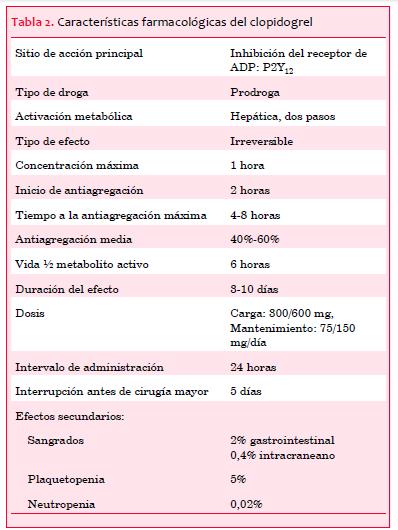
Varios estudios en diferentes escenarios han demostrado que clopidogrel asociado al AAS es superior a la monoterapia con aspirina, reduciendo morbilidad y mortalidad. Esto se aplica a los síndromes coronarios agudos en los que la doble terapia antiplaquetaria (DAP), es decir, AAS más clopidogrel, constituye el tratamiento antiplaquetario estándar.
En el estudio CURE, en pacientes con SCAsST con biomarcadores cardíacos elevados o infradesnivel del segmento ST, tratados con AAS, el clopidogrel (dosis carga de 300 mg + 75 mg de mantenimiento) durante 9-12 meses, redujo la incidencia de muerte cardiovascular, de IAM no fatal o ataque cerebrovascular, comparado con AAS como monoterapia (9,3% versus 11,4%, RR: 0,80, CI95%, 0,72-0,90, p<0,001). Este beneficio se observó en todos los subgrupos de pacientes y fue mantenido durante todo el período de seguimiento, tanto en los tratados en forma conservadora como en los sometidos a tratamiento intervencionista(20,21). Si bien se comprobó un aumento en la incidencia de sangrado mayor con clopidogrel (3,7% versus 2,7%, RR: 1,38, CI95%: 1,13-1,67, p=0,001), no hubo aumento de sangrados fatales, de tal manera que el beneficio clínico neto favorece el uso de clopidogrel sumado a AAS.
La pieza de evidencia más relevante respecto al esquema de administración de clopidogrel, luego de los estudios CURE y PCI CURE, probablemente sea el estudio CURRENT-OASIS 7. En este estudio, además de la comparación de dosis bajas y altas de AAS ya referida, se comparó la utilización de dosis de clopidogrel “altas” (600 mg carga + 150 mg por 7 días y luego 75 mg/día) versus “bajas” (300 mg carga + 75 mg/día) en pacientes con SCA agudo sin y con sobreelevación del segmento ST. Si bien en la población global no se comprobó diferencias entre ambas dosis, un análisis del subgrupo de pacientes tratados con angioplastia coronaria (17.263 pacientes, 63% con SCAsST) demostró una reducción significativa del objetivo primario combinado de muerte cardiovascular/IAM/ataque cerebrovascular (3,9% versus 4,5%, HR: 0,86, IC95%: 0,74-0,99, p=0,039), debido a una reducción de la tasa de IAM en el régimen de dosis elevada. La tasa de trombosis intrastent también se redujo significativamente, independientemente del tipo de stent. No se comprobó diferencia en la incidencia de sangrado fatal o intracraneal.
En pacientes con SCAcST tratados con fibrinolíticos y AAS, los estudios CLARITY y COMMIT evaluaron el uso asociado de clopidogrel. Se comprobó una reducción significativa en los objetivos primarios (mortalidad e infarto) a favor del agregado de clopidogrel a la aspirina(22,23 ), lo que constituye la base del uso de clopidogrel en los SCAcST sometidos a tratamiento fibrinolítico.
El clopidogrel debe ser administrado tan pronto como sea posible. La precocidad de su administración se asocia con mayor eficacia en caso de angioplastia coronaria. Es así que el estudio CREDO demostró que el beneficio de clopidogrel en pacientes sometidos a angioplastia coronaria es evidente cuando la dosis carga de clopidogrel es administrada más de 6 horas antes de la angioplastia(24). Esto es aplicable a los SCAsST, dado que en los SACcST los plazos de reperfusión no permiten cumplir este requisito.
Como dato de interés, un subgupo de análisis del estudio CURRENT-OASIS 7 mostró una reducción de la incidencia de trombosis del stent cuando se utiliza dosis carga de clopidogrel de 600 mg + 150 mg de mantenimiento por seis días y posteriormente 75 mg/día respecto a dosis de mantenimiento convencionales(12).
Finalmente, el estudio HORIZONS-AMI demostró que la dosis carga de clopidogrel 600 mg disminuyó mortalidad, infarto, reinfarto y trombosis del stent a 30 días de forma significativa. El análisis multivariado mostró que la carga de 600 mg se asoció de forma independiente a la reducción de eventos cardiovasculares mayores (MACE, HR 0,72, p=0,04)(25). En voluntarios sanos, la dosis carga de 300 mg de clopidogrel produce un rápido efecto antiplaquetario, con niveles de inhibición cercanos a los logrados en estado estacionario. Las dosis subsiguientes de 75 mg/día mantienen este nivel de inhibición. A pesar de esto, un estudio en pacientes con enfermedad arterial coronaria mostró que solo 16% de los pacientes alcanzan una inhibición plaquetaria mayor a 70%, dos horas luego de una dosis carga de 600 mg. Estos hallazgos sugieren que la respuesta al clopidogrel puede ser variable, lo que constituye uno de los puntos débiles de este fármaco(26,27).
La respuesta disminuida al clopidogrel se asocia con un riesgo incrementado de eventos trombóticos, mientras que una respuesta incrementada se asocia a mayor riesgo de sangrado(2). Las razones para esta variabilidad son múltiples. Existen factores clínicos involucrados que incluyen: una alta reactividad plaquetaria pretratamiento, diabetes (que puede incrementar la reactividad plaquetaria), cumplimiento irregular de la medicación o interacción con otros fármacos. También pueden incidir factores catalogados como celulares, entre los que se incluyen: el incremento del turn over plaquetario y la sobreestimulación de las vías de activación plaquetaria. La respuesta a clopidogrel también es afectada por la variabilidad genética de las enzimas encargadas de su metabolismo hepático. Se han descrito variantes como las del alelo CYP2C19*2, que se asocian significativamente a mayor riesgo de experimentar eventos cardiovasculares mayores, muerte o trombosis del stent. Otra desventaja del clopidogrel consiste en el carácter irreversible de su unión al receptor P2Y12, lo que incrementa el riesgo de sangrado en pacientes que requieren cirugía de revascularización miocárdica de urgencia, en lo que se basa la recomendación de suspender el clopidogrel cinco días antes de la cirugía. Sin embargo, este período libre de tratamiento con clopidogrel en el contexto de un SCA reciente se asocia a un riesgo de IAM cercano a 1%(28).
Se ha ensayado sin éxito incrementar la dosis de clopidogrel para superar la variabilidad interindividual. El estudio Gravitas no comprobó beneficio asociado a duplicar la dosis de clopidogrel en pacientes con SCA en los que se había demostrado resistencia al clopidogrel a través de una actividad plaquetaria residual elevada. Esto no debe confundirse con el resultado positivo del estudio CURRENT- OASIS 7 que logró demostrar beneficio del uso de doble dosis de clopidogrel en la población global (no seleccionada por reactividad plaquetaria) durante la primera semana de tratamiento en pacientes con SCA sometidos a angioplastia coronaria.
En línea con los resultados del estudio Gravitas, no existe evidencia contundente sobre el beneficio de realizar búsqueda de polimorfismos para CYP2C19 o de realizar test de reactividad plaquetaria para ajustar el tratamiento(2).
El clopidogrel tiene un buen perfil de seguridad, dado que no se ha comprobado incremento significativo de sangrados respecto a aspirina como monoterapia. El 1% de los pacientes presenta efectos adversos siendo la reacción alérgica cutánea la más frecuente.
Existe controversia respecto a la interacción farmacológica entre omeprazol y clopidogrel. Si bien existe fundamento teórico para esta interacción con potencial disminución del efecto de clopidogrel, los estudios clínicos no han demostrado asociación con eventos cardiovasculares adversos. Con mucha frecuencia, los inhibidores de la bomba de protones se prescriben de forma simultánea con la DAP, dado que son más eficaces que los agentes anti H2 para prevenir el sangrado gastrointestinal en este contexto(29). Existe información contradictoria acerca de una posible interacción entre estos fármacos en el sentido de que la administración simultánea de ambos puede disminuir el efecto antiagregante del clopidogrel. Esta interacción ha sido documentada midiendo la agregabilidad plaquetaria residual como ocurrió en el estudio PRINCIPLE TIMI 44(30). Tras la administración de 600 mg de clopidogrel, la inhibición de la agregación plaquetaria a las seis horas fue inferior en pacientes tratados con inhibidores de la bomba de protones comparado con los no tratados (p=0,02). Sin embargo, esta diferencia no se traduce en una mayor incidencia de eventos adversos. En el estudio TRITON TIMI(38), el 33% de los pacientes recibió un inhibidor de la bomba de protones. No se puso en evidencia una asociación entre el objetivo cardiovascular combinado del estudio y la administración concomitante de inhibidores de la bomba de protones y clopidogrel o prasugrel. En el estudio COGENT, una población de pacientes con indicación de doble terapia antiagregante con AAS y clopidogrel, se asignó de manera aleatoria a omeprazol o placebo(31). No se comprobó diferencia en la incidencia de eventos cardíacos adversos, pero sí una mayor incidencia de sangrado entre los pacientes no tratados con omeprazol.
En resumen, se acepta utilizar inhibidores H2 en pacientes como estrategia básica de protección gástrica y reservar los inhibidores de la bomba de protones (preferentemente no omeprazol) para los pacientes con riesgo elevado de sangrado (60 años, sangrado previo, ulcus péptico, H. pylori, tratamiento con corticoides, anticoagulación concomitante). El pantoprazol podría constituir una opción preferible al omeprazol, con menor interacción farmacológica con clopidogrel. De todas formas se considera que este no es un problema totalmente resuelto y se necesita mayor evidencia al respecto(32).
2.b.I.b. Prasugrel
El prasugrel es una tienopiridina de administración oral. Se absorbe rápidamente a nivel intestinal y es hidrolizado por la caroxilesterasa 2 a tiolactona R-95913 (compuesto intermedio). En el caso de prasugrel, a diferencia de clopidogrel, la esterasa juega un rol esencial en la activación de la pro droga. El compuesto R-95913 es posteriormente metabolizado por las enzimas CYP3A, CYP2B6, CYP2C9 y CYP2C19 a su metabolito activo R-138727 en un solo paso metabólico a nivel hepático(33).
En comparación con clopidogrel, el prasugrel es más rápido y más potente, logrando concentraciones más elevadas de metabolismo activo. Su concentración máxima se alcanza a los 30 minutos poscarga. Ha demostrado ser superior en todas las fases del tratamiento comparándolo a dosis carga variables de clopidogrel e incluso doble dosis de mantenimiento. La respuesta de prasugrel no solo es más intensa en la magnitud de la inhibición plaquetaria, sino también más predecible(2). Las principales características farmacológicas del prasugrel se resumen en la tabla 3.
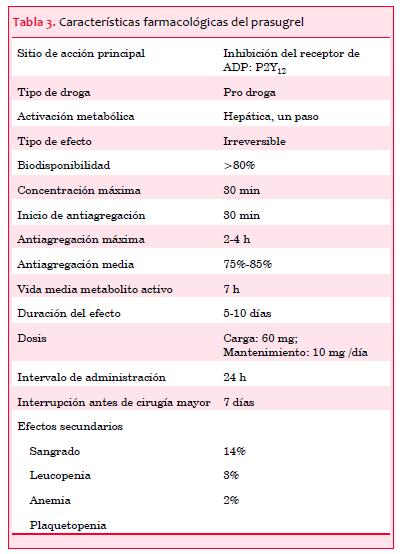
El estudio TRITON-TIMI 38 (13.608 pacientes) comparó clopidogrel versus prasugrel en pacientes con SCA de riesgo moderado y alto en que se planificó una conducta intervencionista. El prasugrel produjo una disminución en el objetivo primario (muerte de causa cardiovascular, IAM no fatal y ACV no fatal) en comparación con dosis estándar de clopidogrel durante 6-15 meses (9,9% versus 12,1%, HR 0,81, p<0,001), a expensas de la reducción en IAM no fatal. También se comprobó el beneficio del prasugrel en relación con clopidogrel respecto a objetivos secundarios como necesidad de nueva revascularización urgente del vaso tratado (HR 0,66, p<0,001) y trombosis del stent (HR 0,48, p<0,001)(34). Un análisis poshoc del estudio TRITON-TIMI 38 mostró que no hubo beneficio en tres subgrupos de pacientes: mayores de 75 años, con ACV o accidente isquémico transitorio previo y bajo peso (menos de 60 kg). Por otro lado, también se demostró que la superioridad de prasugrel respecto a clopidogrel es mayor en pacientes que cursan un IAM con elevación del ST que van a angioplastia, en pacientes diabéticos y en pacientes que requieren cirugía de revascularización miocárdica.
En base a esta evidencia, el prasugrel se encuentra aprobado para su uso en SCA en pacientes con indicación de procedimiento coronario intervencionista en dosis de 60 mg como dosis carga y 10 mg como dosis de mantenimiento, asociado a AAS 75-325 mg.
En cuanto a su perfil de seguridad, prasugrel exhibe mayor riesgo de sangrado respecto a clopidogrel. El riesgo de sangrado fue 2,4% versus 1,8% (HR 1,32, p=0,03), que también incluyó sangrado con riesgo vital 1,4% versus 0,9% (HR 1,52, p=0,01). El riesgo en pacientes que van a cirugía cardíaca es aun mayor: 14,1% versus 4,5%, en especial si la cirugía es realizada dentro de los tres días de la administración de la tienopiridina (26,7% versus 5,0%) y menor si la cirugía se realiza entre cuatro y siete días de la administración. Por esta razón se recomienda la suspensión de prasugrel siete días antes de la cirugía y no administrarlo antes de conocer la anatomía lesional coronaria(34).
A diferencia de clopidogrel, para prasugrel no hay potencial interacción con los inhibidores de la bomba de protones. Los polimorfismos de la CYP2C19 o ABCB1 no afectan al prasugrel. La incidencia de reacciones alérgicas fue menor a 1% en el estudio TRITON –TIMI 38(34).
2.b.II. Inhibidores de los receptores P2Y12 no tienopiridínicos: ticagrelor
El ticagrelor pertenece a una nueva clase de agentes, las ciclopentiltriazolopirimidinas (CPTPs). El ticagrelor se une de forma reversible y no competitiva al receptor P2Y12, lo que probablemente ocurra en un sitio de unión diferente al de las tienopiridinas. No es una pro droga, por lo que no requiere activación metabólica alguna. Es metabolizado por la enzima CYP3A y uno de sus metabolitos activos (AR-C124910XX) alcanza concentraciones de aproximadamente 40% de la de ticagrelor en plasma y tiene igual potencia de acción que este.
Ticagrelor se absorbe rápidamente luego de administración oral y alcanza sus máximas concentraciones en plasma en aproximadamente 1,5 a 3 horas.
Este fármaco ha demostrado mayor potencia y eficacia que clopidogrel con una inhibición plaquetaria superior y más consistente(35). Adicionalmente, la inhibición plaquetaria asociada con ticagrelor es más rápida que con clopidogrel, 30 minutos luego de la dosis carga la inhibición de la actividad plaquetaria fue de 41% para ticagrelor y de 8% para clopidogrel (p<0,0001), en tanto que a los dos horas era de 88% versus 38% respectivamente (p<0,0001). Las características farmacológicas más relevantes del ticagrelor se resumen en la tabla 4.
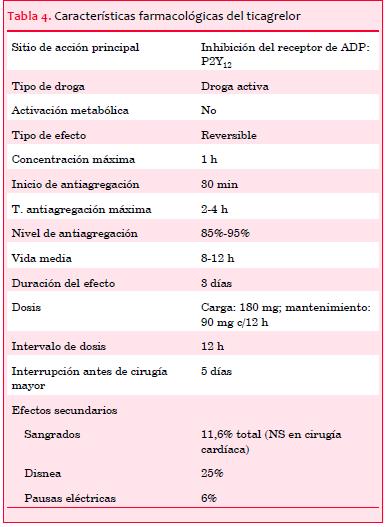
En el estudio PLATO se randomizaron 18.624 pacientes con SCA a tratamiento con ticagrelor (180 mg carga + 90 mg c/12 horas mantenimiento) o clopidogrel (300-600 mg carga + 75 mg/día mantenimiento). Se demostró una disminución significativa en el endpoint primario (muerte de causa cardiovascular, IAM o ACV) en el brazo ticagrelor (9,8% versus 11,7%, HR 0,84, p=0,001), a expensas de la reducción de la incidencia de muerte de causa vascular (4,0% versus 5,1%, p=0,001) e IAM (5,8% versus 6,9%, p=0,005). También se comprobó una reducción significativa de la mortalidad de cualquier causa, IAM o ACV y en el endpoint secundario compuesto de muerte de causa vascular, IAM, ACV, isquemia recurrente severa, isquemia recurrente, ataque cerebrovascular transitorio u otro evento arterial trombótico (14,6% versus 16,7%, HR 0,88, p<,001), pero no hubo diferencia en ACV aislado(35). Estos resultados fueron comprobados en forma consistente en 30 subgrupos analizados poshoc (pacientes que van a intervencionismo, a cirugía cardíaca, ACV o ataque isquémico transitorio previo, enfermedad renal crónica y diabetes mellitus)(36-38). Sin embargo, se observó una menor eficacia en tres subgrupos de pacientes: pacientes con mediana de peso menor a la esperada para su sexo, pacientes que no tomaban hipolipemiantes orales previo a la randomización y pacientes enrolados en centros de América del Norte. Este último punto puede tener que ver con las dosis de AAS utilizadas, superiores a las que se usan en Europa, si bien no se puede descartar que se deba al azar(39).
El polimorfismo genético en las enzimas CYP2C19 y ABCB no afectó la eficacia de ticagrelor a diferencia de clopidogrel y esto se debe a que ticagrelor no requiere activación metabólica.
Tal vez el valor agregado más importante del ticagrelor es que su eficacia es superior a clopidogrel sin incrementar el riesgo de sangrado mayor. No se demostró diferencia significativa en sangrado mayor entre los grupos clopidogrel y ticagrelor en el estudio PLATO(35). Cuando se consideran sangrados mayores y menores en conjunto, el riesgo es mayor para ticagrelor (16,1% versus 14,6%, HR 1,11, p=0,008).
Llamativamente, el síntoma disnea fue significativamente mayor en los pacientes tratados con ticagrelor (13,8% versus 7,8%, HR 1,84, p<0,001), así como también las pausas ventriculares de hasta 3 s durante la primera semana de tratamiento (5,8% versus 3,6%, p=0,01), pero no al día 30 de seguimiento. No obstante, la incidencia de síncope, bradicardia, bloqueo auriculoventricular e implante de marcapaso fueron similares en ambos grupos. La tasa de discontinuación del tratamiento por efectos adversos fue superior para ticagrelor que para clopidogrel (7,4% versus 6,0%, p<0,001), la disnea fue causa de suspensión en 0,9% con ticagrelor versus 0,1% con clopidogrel(35).
Las recomendaciones actuales sugieren suspender ticagrelor cinco a siete días antes de una cirugía electiva.
Son contraindicaciones para ticagrelor la presencia de sangrado patológico activo, historia de hemorragia intracraneal previa y disfunción hepática severa. Se recomienda que la dosis de mantenimiento de AAS no supere los 100 mg/día.
Dentro de las advertencias sobre el uso de ticagrelor figuran las interacciones farmacológicas con inhibidores potentes de la CYP3A4 (ketoconazol) y el uso concomitante con inductores de la CYP3A4 (comitoína). La simvastatina no debe administrarse en dosis superiores a 40 mg/día en concomitancia con ticagrelor, no habiendo interferencia con atorvastatina o rosuvastatina. En caso de tratamiento simultáneo con digoxina, los niveles de esta última deben ser monitorizados debido a su posible aumento.
También se ha visto un discreto incremento en los niveles séricos de creatinina, por lo que se sugiere monitorizar la creatininemia al mes de inicio del ticagrelor, sobre todo en mayores de 75 años y en caso de tratamiento concomitantemente con inhibidores de la enzima conversora de angiotensina (IECA) o antagonistas del receptor de angiotensina II (ARAII). Dado que el uso de ticagrelor puede aumentar la uricemia, se contraindica su uso en pacientes con nefropatía por ácido úrico y se recomienda su uso cauteloso en pacientes con elevación previa de ácido úrico o gota(2).
2.c. Inhibidores de la glicoproteína IIb/IIIa
Los inhibidores GP IIb/IIIa representan otra estrategia en la inhibición de la activación plaquetaria mediada por fibrinógeno. Hay varios agentes de esta clase que cuentan con aprobación para uso intravenoso. No se ha podido desarrollar un fármaco eficaz de esta clase para uso vía oral, existiendo evidencia negativa sobre su utilidad, incluyendo incremento de la mortalidad(40). El uso de estos fármacos está limitado a pacientes que cursan un SCA con indicación de tratamiento intervencionista. El abciximab es un fab (fragment antigen-binding) quimérico de un anticuerpo monoclonal que se une de forma no específica al receptor plaquetario GPIIb/IIIa. Este fármaco ha demostrado disminuir el número de eventos en comparación a placebo en pacientes con SCA sin elevación del ST de alto riesgo que van a tratamiento intervencionista bajo clopidogrel, aunque este beneficio se observa solamente en pacientes con altos niveles de troponina(41). Un metaanálisis que incluyó estudios de pacientes con SCA con elevación del ST que van a angioplastia primaria, mostró beneficio del uso de abciximab en relación con placebo; sin embargo, también hubo incremento en el sangrado. Por tanto, se aconseja su uso solamente en pacientes con SCA de alto riesgo y con biomarcadores positivos.
Otro de los compuestos aprobados es el tirofibán. Se trata de una pequeña molécula no peptídica, antagonista del receptor GPIIb/IIIa, que ha sido extensamente estudiada en el contexto de procedimientos de cardiología intervencionista. El tirofibán ha demostrado ser más efectivo que placebo en la reducción de mortalidad (OR 0,68, p=0,001) y muerte e infarto (OR 0,69, p<,001) a 30 días(42).
El eptifibatide es un heptapéptido cíclico antagonista del receptor GP IIb/IIIa, con una alta especificidad para este receptor de forma reversible. La evidencia a favor de este fármaco es débil pero favorable. Recientemente se ha demostrado que la administración temprana de eptifibatide no presentó ventajas respecto a la administración posangiográfica, lo que soporta la administración ad hoc de inhibidores de la GP IIb/IIIa en pacientes con SCA que van a intervencionismo(43).
3. Aplicación clínica de la antiagregación plaquetaria en los SCA
3.a. Antiplaquetarios en los SCAsST
Tanto las guías europeas como las norteamericanas recomiendan el uso de AAS tan precozmente como sea posible (indicación clase IA)(9,44). Ambas guías también coinciden en la indicación de por vida una vez superado el cuadro agudo, aunque difieren en la dosis. El Colegio americano de Cardiología y la Sociedad Americana de Cardiología (ACC/AHA) recomienda una dosis de 162-325 mg/día o 75-162 mg/día en pacientes con alto riesgo de sangrado, mientras que la guía europea recomienda una dosis de mantenimiento de 75-100 mg/día. Existe evidencia que avala el uso de bajas dosis de mantenimiento de AAS.
La indicación de DAP es formal en los SCAsST de alto riesgo. La guía norteamericana y la europea coinciden en recomendar la administración tan temprana como sea posible de inhibidores del receptor P2Y12 (IA). La guía norteamericana recomienda una dosis carga de clopidogrel de 600 mg previo a intervencionismo, con una dosis de 75 mg de mantenimiento (indicación IB), o ticagrelor en dosis carga de 180 mg y dosis de mantenimiento de 90 mg cada 12 horas (indicación IB). También se recomienda con el mismo nivel de evidencia la utilización de prasugrel. Se recomienda solo luego de conocer la anatomía coronaria y luego de tomar la decisión de realizar angioplastia coronaria. La dosis de prasugrel es de 60 mg carga y 10 mg/día mantenimiento, con ajuste a 5 mg/día en pacientes mayores a 75 años o peso menor a 60 kg (IB).
La guía europea, sin embargo, ubica a prasugrel (solo luego de conocer la anatomía coronaria) y a ticagrelor por encima de clopidogrel, teniendo en cuenta los resultados de los estudios TRITON TIMI-38 y PLATO (IB), y reserva el uso de clopidogrel solo para la falta de disponibilidad de ticagrelor o prasugrel (IC).
Ambas guías también recomiendan la administración de doble dosis de clopidogrel en la primera semana pasando luego a 75 mg/día de mantenimiento, aunque con diferente categoría en el nivel de recomendación (IIb B para la AHA/ACC y IIa B para la Sociedad Europea de Cardiología [ESC]).
En pacientes en que se opta por tratamiento primariamente conservador, la guía norteamericana recomienda clopidogrel o ticagrelor (IB), mientas que la guía europea recomienda ticagrelor en primer lugar (IB) y clopidogrel en segundo lugar (IC).
Ambas guías coinciden en que el tiempo de doble antiagregación debe ser de un año (IB).
De acuerdo a las guías ACC/AHA, los inhibidores de la GP IIb/IIIa, preferentemente eptifibatide o tirofibán (IB), pueden ser utilizados antes del procedimiento de cardiología intervencionista cuando los pacientes no recibieron inhibidores del receptor P2Y12 (IA), siendo esta recomendación de menor nivel en la guía europea (IIa C). También se recomienda el uso de tirofibán o eptifibatide en pacientes que han recibido AAS, clopidogrel o ticagrelor y un anticoagulante y son referidos a angiografía y eventual angioplastia, siempre que el riesgo de sangrado sea bajo (ACC/AHA: IIa C, ESC: IIb C).
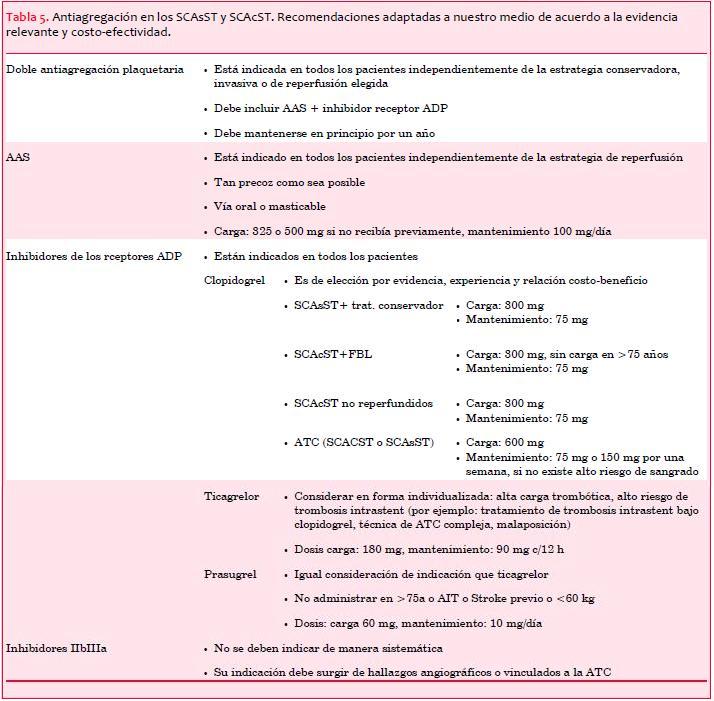
Desde el punto de vista práctico creemos conveniente realizar recomendaciones adaptadas a nuestro medio en cuanto a experiencia, evidencia y relación costo-beneficio sobre el tratamiento antiagregante en los SCA, que se presentan en la tabla 5 en conjunto para los SCAsST como para los SCAcST.
3.b. Antiplaquetarios en los SCAcST
El beneficio del AAS en el SCAcST ha sido claramente establecido. En el estudio ISIS-2, 17.187 pacientes con sospecha de IAM fueron randomizados a aspirina, estreptoquinasa, ambos o placebo; la aspirina redujo la mortalidad vascular en 23% sin incrementar significativamente el riesgo de sangrado y este resultado fue de similar magnitud y aditivo al de la estreptoquinasa(45). Este beneficio fue confirmado además mediante un metaanálisis que incluyó a 212.000 pacientes(11). Por lo tanto, la indicación de AAS es formal, tan precozmente como sea posible, con formulación masticable de preferencia. La recomendación del uso temprano de AAS es IB tanto en la guía norteamericana como en la europea.
Existen diferencias en la dosis recomendada (AHA/ACC: 162-325 mg, ESC: 150-300 mg oral o 80-150 mg por vía intravenosa). Las dosis de mantenimiento son también diferentes, al igual que en las guías de SCA sin ST (ACC/AHA: 162-325 mg, ESC: 75-100 mg). En todos los casos se debe mantener el tratamiento de por vida (IA). En realidad no hay diferencia en la rapidez y magnitud de la antiagregación con estas diferencias de dosis. Sin embargo, de acuerdo a los hallazgos del estudio PLATO respecto a diferencias regionales en la efectividad de ticagrelor, es razonable utilizar dosis de AAS no mayores de 100 mg para evitar una posible interferencia entre AAS y ticagrelor(46 ).
En cuanto a los inhibidores del receptor P2Y12 se recomienda utilizarlos precozmente y con dosis carga. La guía norteamericana no tiene preferencia entre clopidogrel, prasugrel y ticagrelor (IB), mientras que la guía europea pone por encima a prasugrel (en pacientes que no han recibido clopidogrel y que son menores de 75 años) y ticagrelor (IB) respecto a clopidogrel (que se debería utilizar solo cuando no hay disponibilidad de prasugrel o ticagrelor, IC). La dosis de clopidogrel varía de acuerdo a la estrategia de reperfusión aplicada. En caso de angioplastia coronaria primaria valen las dosis antes referidas (carga 600 mg y mantenimiento 75/150 mg/día). En caso de tratamiento fibrinolítico, la dosis carga será de 300 mg en menores de 75 años y no se indicará dosis carga a mayores de 75 años. En SCAcST no reperfundidos se utilizará igual esquema de dosificación de clopidogrel.
La duración de la doble antiagregación es de 12 meses para ambas guías (IB). Realizaremos consideraciones adicionales sobre la duración de la doble terapia antiagregante a propósito de doble antiagregación y anticoagulación.
La utilización de rutina de inhibidores de la GP IIb/IIIa durante el intervencionismo es catalogada como razonable en ambas guías, pero se ha reducido su importancia en comparación con guías previas cuando se utiliza heparina no fraccionada como anticoagulante (ACC/AHA: IIaB, ESC: IIbB). Durante el intervencionismo el abciximab es recomendación IA en ambas guías, respecto a doble bolo de eptifibatide o bolo de alta dosis de tirofibán (IB). La guía europea también plantea la utilización de inhibidores de la GP IIb/IIIa frente a una alta carga trombótica o fenómeno de no reflujo o flujo lento (IIa C).
La diferencia más importante entre la guía europea y la norteamericana consiste en la recomendación de ticagrelor y prasugrel por encima de clopidogrel, basada solamente en la existencia de un estudio para cada comparación: PLATO para ticagrelor y TRITON TIMI-38 para prasugrel. Existe actualmente debate sobre la objetividad de la recomendación europea de superioridad de ticagrelor y prasugrel respecto a clopidogrel(47).
Si bien la opinión de la comunidad científica respecto a los estudios PLATO y TRITON TIMI-38 es mayoritaria respecto a la superioridad de ticagrelor y prasugrel en relación con clopidogrel, parece razonable ser cautos y esperar mayor volumen de evidencia. El clopidogrel es un fármaco seguro y de gran eficacia, comprobado en numerosos estudios randomizados y avalado por la experiencia.
Recomendaciones adaptadas a nuestro medio sobre el tratamiento antiagregante en los SCAcST se presentan en la tabla 5 en conjunto con las correspondientes a los SCAsST.
3.c. Riesgo de discontinuación de la doble antiagregación
La discontinuación prematura de la DAP incrementa significativamente el riesgo de eventos cardíacos adversos mayores (MACE) luego de cualquier angioplastia y en especial luego de un SCA. La duración de la doble antiagregación es controvertida y depende del contexto clínico y del tipo de stent. En el caso de los SCA la recomendación más generalizada es mantener la DAP por un año, independientemente del tipo de stent implantado. El predictor independiente más importante de trombosis intrastent precoz y tardía es la ausencia de tratamiento con inhibidores P2Y12(48). De todas formas, no toda discontinuación de la DAP tiene igual riesgo. Existe evidencia reciente que indica que cuando la discontinuación es debida a prescripción médica en un contexto de estabilidad del paciente, el riesgo de MACE es muy bajo(49). Por otro lado, cuando la interrupción ocurre ya sea por voluntad unilateral del paciente o por alguna condición médica que contraindica la antiagregación, como un sangrado en curso, el riesgo de MACE, en especial de trombosis intrastent, es elevado. Además, debe tenerse en cuenta que el período de mayor riesgo es la semana siguiente a la suspensión (siete veces más), persistiendo elevado hasta los 30 días aproximadamente. Vale la pena mencionar que 80% de todas las trombosis ocurre bajo medicación, lo que se atribuye a la mejoría en el diseño de las plataformas de los stents que hace menos probable la trombosis aun luego de suspendida la DAP. Cuando el paciente suspende unilateralmente la medicación, la trombosis del stent tiene una incidencia de 14% aproximadamente. Cuando es el médico quien de forma planificada suspende definitiva o transitoriamente la medicación, el riesgo es bajo. Estos datos sugieren que el criterio clínico del cardiólogo tratante es una variable de alto valor que se asocia al éxito o al fracaso de la terapia antiplaquetaria en la prevención de MACE.
Bibliografía
1. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007;357:2482-94.
2. Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012;72:2087-116.
3. Dorsam RT, Kunapuli SP. Central role of the P2Y12 receptor in platelet activation. J Clin Invest 2004;113:340-5.
4. Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res 2006;99:1293-304.
5. Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation 2006;114:1070-7.
6. Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010;74:597-607.
7. Wright RS, Anderson JL, Adams CD, Bridges CR, Casey DE Jr, Ettinger SM, et al. 2011 ACCF/AHA focused update of the guidelines for the management of patients with unstable angina/ non-ST-elevation myocardial infarction (Updating the 2007 guideline): a report of the American College Of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2011;123:2022-60.
8. Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, Cercek B, et al. 2011 ACCF/ AHA/SCAI guideline for percutaneous coronary intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011;124:e574-651.
9. Hamm CW, Bassand JP, Agewall S, Bax J, Boersma E, Bueno H, et al. Esc guidelines for the management of acute coronary syndromes in patients presenting without persistent st-segment elevation: the Task Force For The Management of acute coronary syndromes (ACS) in patients presenting without persistent ST-Segment elevation of the European Society Of Cardiology (ESC). Eur Heart J 2011;32:2999-3054.
10. Wijns W, Kolh P, Danchin N, Di Mario C, Falk V, Folliguet T, et al. Guidelines on myocardial revascularization. Eur Heart J 2010;31:2501-55.
11. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002;324(7329):71-86.
12. Mehta SR, Bassand JP, Chrolavicius S, Diaz R, Eikelboom JW, Fox KA, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010;363:930-42.
13. Vachhani R, Bouhaidar D, Zfass A, Sandhu B, Nawras A. Critical appraisal of a fixed combination of esomeprazole and low dose aspirin in risk reduction. Ther Clin Risk Manag 2010;6:287-92.
14. Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018-24.
15. Peters RJG, Mehta SR, Fox KAA, Zhao F, Lewis BS, Kopecky SL, et al. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: Observations from the Clopidogrel In Unstable Angina To Prevent Recurrent Events (CURE) study. Circulation 2003; 108(14):1682-7.
16. Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Huisman MV. Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch Intern Med 2007;167: 1593-9.
17. Collet JP, Montalescot G, Blanchet B, Tanguy ML, Golmard JL, Choussat R, et al. Impact of prior use or recent withdrawal of oral antiplatelet agents on acute coronary syndromes. Circulation 2004;110:2361-7.
18. Storey RF. Biology and pharmacology of the platelet P2Y12 receptor. Curr Pharm Des 2006;12:1255-9.
19. Lins R, Broekhuysen J, Necciari J, Deroubaix X. Pharmacokinetic profile of 14C-labeled clopidogrel. Semin Thromb Hemost 1999;25 Suppl 2:29-33.
20. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001;345(7):494-502.
21. Mehta SR, Yusuf S, Peters RJG, Bertrand ME, Lewis BS, Natarajan MK, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001;358(9281):527-33.
22. Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005;352:1179-89.
23. Chen ZM, Jiang LX, Chen YP, Xie JX, Pan HC, Peto R, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366: 1607-21.
24. Steinhubl SR, Berger PB, Mann JT 3rd, Fry ET, DeLago A, Wilmer C, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: A randomized controlled trial. JAMA 2002;288:2411-2420.
25. Dangas G, Mehran R, Guagliumi G, Caixeta A, Witzenbichler B, Aoki J, et al. Role of clopidogrel loading dose in patients with st-segment elevation myocardial infarction undergoing primary angioplasty: results from the HORIZONS-AMI (Harmonizing Outcomes With Revascularization and stents in acute myocardial infarction) trial. J Am Coll Cardiol 2009;54:1438-46.
26. Savcic M, Hauert J, Bachmann F, Wyld PJ, Geudelin B, Cariou R. Clopidogrel loading dose regimens: kinetic profile of pharmacodynamic response in healthy subjects. Semin Thromb Hemost 1999;25 Suppl 2:15-9.
27. Gurbel PA, Bliden KP, Butler K, Tantry US, Gesheff T, Wei C, et al. Randomized double-blind assessment of the onset and offset of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the onset/offset study. Circulation 2009;120:2577-85.
28. Kunadian B, Thornley AR, Tanos M, Dunning J. Should clopidogrel be stopped prior to urgent cardiac surgery? Interact Cardiovasc Thorac Surg. 2006;5(5):630-6.
29. Fook-Hong N, Wong S-Y, Lam K-F, Chu W-M, Pierre C, Ling Y-H, et al. Famotidine is inferior to pantoprazole in preventing recurrence of aspirin-related peptic ulcers or erosions. Gastroenterology 2010;138:82-8.
30. Wiviott SD, Trenk D, Frelinger AL, O’Donoghue M, Neumann F-J, Michelson AD, et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the prasugrel in comparison to Clopidogrel for inhibition of platelet activation and aggregation–thrombolysis in myocardial infarction PRINCIPLE-TIMI 44 trial. Circulation 2007;116:2923-32.
31. Bhatt DL, Cryer BL, Contant CF, Cohen M, Lanas A, Schnitzer TJ, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med 2010;363:1909-17.
32. Bhatt DL, Cryer BL, Contant CF, Cohen M, Lanas A, Schnitzer TJ, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med 2010;363:1909-17.
33. Farid NA, Kurihara A, Wrighton SA. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J Clin Pharmacol 2010;50:126-42.
34. Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007;357:2001-15.
35. Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009;361:1045-57.
36. James SK, Storey RF, Khurmi NS, Husted S, Keltai M, Mahaffey KW, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes and a history of stroke or transient ischemic attack. Circulation 2012;125:2914-21.
37. James S, Budaj A, Aylward P, Buck KK, Cannon CP, Cornel JH, et al. Ticagrelor versus clopidogrel in acute coronary syndromes in relation to renal function: Results from the platelet inhibition and patient outcomes (PLATO) trial. Circulation 2010;122:1056-67.
38. James S, Angiolillo DJ, Cornel JH, Erlinge D, Husted S, Kontny F, et al. Ticagrelor vs. Clopidogrel in patients with acute coronary syndromes and diabetes: a substudy from the platelet inhibition and patient outcomes (PLATO) trial. Eur Heart J 2010;31:3006-16.
39. Mahaffey KW, Wojdyla DM, Carroll K, Becker RC, Storey RF, Angiolillo DJ, et al. Ticagrelor compared with clopidogrel by geographic region in the platelet inhibition and patient outcomes (PLATO) trial. Circulation 2011;124:544-54.
40. Chew DP, Bhatt DL, Sapp S, Topol EJ. Increased mortality with oral platelet glycoprotein iib/iiia antagonists: A meta-analysis of phase III multicenter randomized trials. Circulation 2001;103:201-6.
41. Kastrati A, Mehilli J, Neumann FJ, Dotzer F, ten Berg J, Bollwein H, et al. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: The isar-react 2 randomized trial. JAMA 2006;295:1531-8.
42. Valgimigli M, Biondi-Zoccai G, Tebaldi M, van’t Hof AW, Campo G, Hamm C, et al. Tirofiban as adjunctive therapy for acute coronary syndromes and percutaneous coronary intervention: A meta-analysis of randomized trials. Eur Heart J 2010;31:35-49.
43. Giugliano RP, White JA, Bode C, Armstrong PW, Montalescot G, Lewis BS, et al. Early versus delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009;360:2176-90.
44. Jneid H, Anderson JL, Wright RS, Adams CD, Bridges CR, Casey DE, Jr., et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non- st-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645-81.
45. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. ISIS-2 (Second International Study Of Infarct Survival) Collaborative Group. Lancet 1988;2:349-60.
46. Huber K, Lip GY. Differences between ACC/AHA and ESC guidelines on antiplatelet therapy in patients with acute coronary syndromes. Thromb Haemost 2013; 110 (1):11-3.
47. Serebruany VL, Dinicolantonio J. Mismatch between the European and American Guidelines on Oral Antiplatelet P2Y12 inhibitors after acute coronary syndromes. Thromb Haemost 2013; 110(1):5-10.
48. van Werkum JW, Heestermans AA, Zomer AC, Kelder JC, Suttorp M-J, Rensing BJ, et al. Predictors of coronary stent thrombosisthe Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009;53: 1399-409.
49. Mehran R, Baber U, Steg PG, Ariti C, Weisz G, Witzenbichler B, et al. Cessation of dual antiplatelet treatment and cardiac events after percutaneous coronary intervention (Paris): 2 year results from a prospective observational study. Lancet 2013 Aug 30. pii: S0140-6736(13)61720-1.


{kind=link}
{kind=link}