










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Uruguaya de Cardiología
On-line version ISSN 1688-0420
Rev.Urug.Cardiol. vol.27 no.3 Montevideo Dec. 2012
Revisión
Miocardiopatías inducidas por arritmias: ¿el enigma del huevo o la gallina todavía sin resolver?
Emmanuel N. Simantirakis, Emmanuel P. Koutalas, Panos E. Vardas
Reproducido de Europace 2012; 14: 466–73. Traducido al español con autorización
Departamento de Cardiología, Hospital Universitario Heraklion, PO Box 1352, 71110 Heraklion, Creta, Grecia
Recibido el 18 de Julio de 2011; aceptado tras su revisión el 10 de octubre de 2011; publicación en línea el 13 de noviembre de 2011.
Conflicto de interés: no se declara ninguno.
Resumen
Especialmente en las últimas décadas varios estudios han intentado confirmar la hipótesis que sostiene que la contracción miocárdica inapropiadamente rápida, irregular, o asincrónica provoca miocardiopatía. Las frecuencias ventriculares rápidas resultantes de arritmias supraventriculares y fibrilación auricular (FA), la irregularidad del ritmo cardíaco —elemento básico de la FA— y la asincronía, determinada por estimulación ventricular derecha, bloqueo de rama, o complejos ventriculares prematuros frecuentes, han sido establecidas como las causas primarias de miocardiopatía inducida por arritmias. Se han aclarado las principales vías fisiopatológicas involucradas; éstas incluyen la activación neurohumoral, el agotamiento de los depósitos energéticos, y anomalías que ocurren durante el estrés y la sobrecarga. Desgraciadamente, desde un punto de vista clínico, los pacientes habitualmente consultan recién cuando aparecen síntomas, a pesar de que la arritmia causal pueda haber aparecido ya meses o años antes, llevando así a una remodelación del miocardio y disfunción diastólica y sistólica. En algunos casos, tratar de establecer un diagnóstico definitivo puede convertirse en un ejercicio agotador para el médico tratante, ya que la arritmia puede no estar siempre presente, y además, implica que primero tiene que indicar un tratamiento, para confirmar el diagnóstico recién retrospectivamente. Otra dificultad que plantea el proceso diagnóstico es que los criterios diagnósticos estrictos siguen siendo tema de controversia. Las opciones terapéuticas incluyen pruebas empíricas con agentes farmacológicos, terapias por catéter, y en el contexto de la estimulación ventricular crónica, la resincronización. Es de esperar que en la mayoría de los pacientes se logre una recuperación parcial o completa, pero aun así los pacientes tendrán que ser seguidos cuidadosamente debido al riesgo de recurrencia. Se hace entonces indispensable realizar ensayos aleatorizados controlados de gran tamaño que permitan optimizar la estratificación de los pacientes y definir la elección de estrategias terapéuticas.
Palabras clave:
MIOCARDIOPATíA INDUCIDA POR TAQUICARDIA
CARDIOPATíAS REVERSIBLES
ASINCRONíA MIOCáRDICA
MIOCARDIOPATíAS INDUCIDAS POR ARRITMIAS
Summary
The hypothesis testing of inappropriate fast, irregular, or asynchronous myocardial contraction provoking cardiomyopathy has been the primary focus of numerous research efforts, especially during the last few decades. Rapid ventricular rates resulting from supraventricular arrhythmias and atrial fibrillation (AF), irregularity of heart rhythm—basic element of AF—and asynchrony, as a consequence of right ventricular pacing, bundle branch block, or frequent premature ventricular complexes, have been established as primary causes of arrhythmia-induced cardiomyopathy. The main pathophysiological pathways involved have been clarified, including neurohumoral activation, energy stores depletion, and abnormalities in stress and strain. Unfortunately, from a clinical point of view, patients usually seek medical advice only when symptoms develop, while the causative arrhythmia may be present for months or years, resulting in myocardial remodelling, diastolic, and systolic dysfunction. In some cases, making a definite diagnosis may become a strenuous exercise for the treating physician, as the arrhythmia may not be present and, additionally, therapy must be applied for the diagnosis to be confirmed retrospectively. The diagnostic process is also hardened due to the fact that strict diagnosing criteria are still a matter of discrepancy.
Therapy options include pharmaceutical agents trials, catheter-based therapies and, in the context of chronic ventricular pacing, resynchronization. For the majority of patients, partial or complete recovery is expected, although they have to be followed up thoroughly due to the risk of recurrence. Large, randomized controlled trials are more than necessary to optimize patients’ stratification and therapeutic strategy choices.
Key words:
TACHYCARDIA-INDUCED CARDIOMYOPATHIES
REVERSIBLE CARDIOMYOPATHIES
MYOCARDIAL ASYNCHRONY
ARRHYTHMIA INDUCED CARDIOMYOPATHIES
Introducción
Cuando un médico se ve frente a un paciente con una miocardiopatía que incluya alteración de la función auricular y/o ventricular y arritmias crónicas o repetitivas, debe tener en cuenta que lo que tiene frente a sí tal vez no represente un diagnóstico final sino la consecuencia de un mecanismo causal primario de base. ¿Qué vino primero? ¿La miocardiopatía o la arritmia? En las últimas décadas han aparecido importantes evidencias que muestran que casi cualquier forma de arritmia supraventricular sostenida o de tipo repetitivo puede provocar disfunción miocárdica. Datos que surgen de investigación reciente han demostrado que la contracción asincrónica del miocardio (como por ejemplo: complejos ventriculares prematuros frecuentes [CVP], bloqueo de rama, y estimulación ventricular) también son capaces de inducir alteración de la función ventricular. Todos los factores arriba mencionados pueden llevar a una reducción de la fracción de eyección (FE) del ventrículo, dilatación ventricular, y finalmente a una insuficiencia cardíaca congestiva, a través de vías fisiopatológicas que no difieren sustancialmente de las establecidas para una miocardiopatía dilatada. Sin embargo, existe evidencia que demuestra que es posible revertir dichas formas de miocardiopatía, parcial o completamente, siempre y cuando se instauren esquemas de tratamiento apropiados.
Definiciones: necesitan actualización
Hay que reconocer que la terminología utilizada hasta ahora no se aplica a la totalidad de las causas y fenotipos de las miocardiopatías inducidas por taquicardia (MIT). La definición clásica se refiere a una entidad descrita como una alteración de la función del ventrículo izquierdo (VI) secundaria a una taquicardia crónica no controlada, que puede revertir parcial o completamente tras la normalización de la frecuencia cardíaca y/o la eliminación de la irregularidad del ritmo (1). La información actualmente disponible ha llevado a ampliar esta última definición para incluir las disfunciones auriculares y/o ventriculares producidas por frecuencias auriculares o ventriculares elevadas, en ausencia de una enfermedad estructural de base (2). Además, estudios recientes han demostrado que la contracción miocárdica asincrónica, como aquella producida por una CVP o estimulación en la zona apical del ventrículo derecho, puede llevar a una dilatación miocárdica más o menos reversible y síntomas de insuficiencia cardíaca congestiva (3,4). Si uno intentara resumir lo arriba descrito bajo el término de miocardiopatía inducida por arritmia (MIA), estas formas de miocardiopatía podrían describirse como disfunción auricular y/o ventricular secundaria a contracciones miocárdicas rápidas y/o asincrónicas/irregulares, que puede revertir parcial o completamente al tratar la arritmia causal. En nuestra opinión, sería preferible que esa definición no se limitara a los casos en los que no hay enfermedad estructural primaria, ya que los pacientes con una disfunción ventricular conocida también son propensos a sufrir las consecuencias de las MIT (5). Fenelon y colaboradores (6) han dividido las MIT en dos subgrupos: 1) MIT puras, cuando la taquicardia crónica produce lesión del miocardio normal, y ese es el único mecanismo que produce su deterioro, y 2) MIT impuras, en los casos en los que no se cumplen los criterios arriba mencionados.
Presentación clínica y causas
Es difícil estimar la prevalencia, incidencia, y frecuencia de las MIT porque hay pocos estudios publicados; además, los que hay en general son de diseño retrospectivo y abarcan poblaciones pequeñas de pacientes.
La presentación clínica típica es la de un paciente con signos y síntomas de insuficiencia cardíaca congestiva y miocardiopatía dilatada (2). La arritmia causal puede no estar presente en ese momento, por tanto el médico tratante tiene que ser extremadamente cuidadoso al definir el diagnóstico. Además, los pacientes a menudo se presentan recién al cabo de meses o años de aparecida la taquicardia, una vez que los síntomas se hacen evidentes, y a menudo no logran dar información demasiado precisa sobre la fecha de inicio de la arritmia. Por supuesto, no es inhabitual que el diagnóstico surja de los estudios imagenológicos realizados a pacientes que no se quejan de ningún síntoma. Todavía no se ha establecido una frecuencia ventricular crítica por encima de la cual se confirme el diagnóstico de MIT. Sin embargo, es lógico suponer que frecuencias cardíacas >100 min (21) puedan tener consecuencias deletéreas (7).
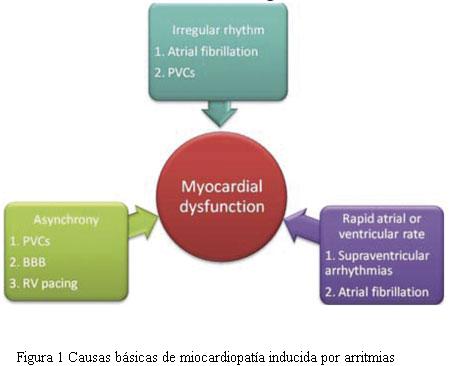
Gossage y colaboradores fueron los primeros en comunicar un caso de un hombre con una miocardiopatía dilatada producida por una fibrilación auricular (FA) rápida (8). Desde entonces, han surgido varios trabajos que demuestran que casi cualquier forma de arritmia supraventricular sostenida puede producir una MIA (figura 1) (9–14). Es preciso aclarar que en pacientes con FA el daño no sólo es producido por las frecuencias ventriculares rápidas; la irregularidad de la respuesta ventricular deteriora aún más la función miocárdica (15). En general, 25%–50% de los pacientes con disfunción del VI y FA presentan un cierto grado de MIT (5,16).
En su estudio pionero de un modelo experimental de MIT, Whipple y colaboradores fueron los primeros en demostrar que la estimulación rápida -ya sea auricular o ventricular- puede inducir alteración de la función auricular o ventricular (13). Los efectos deletéreos de la estimulación apical del ventrículo derecho (VD) han sido atribuidos a un patrón de activación ventricular, eléctrica y mecánica, anormal (17). A la larga, la presencia de disincronía mecánica después de estimulación apical del VD se acompaña de dilatación del VI y deterioro de su función sistólica (18). En la era moderna de ensayos aleatorizados, los estudios de estimulación cardíaca han demostrado que la desincronización ventricular resultante de la estimulación ventricular, aun preservando la sincronización aurículo-ventricular, aumenta el riesgo de hospitalización debido a insuficiencia cardíaca (19). En el ensayo DAVID se distribuyeron aleatoriamente pacientes con una indicación estándar para implantación de desfibrilador, pero sin una indicación de estimulación anti bradicardia; un brazo recibió un marcapasos fisiológico (modo DDDR, con una frecuencia menor a 70 lpm) y el otro un marcapasos de respaldo ventricular (modo VVIR, frecuencia cardíaca inferior a 40 lpm). Las medidas primarias de resultados (ausencia de muerte y ausencia de hospitalizaciones por insuficiencia cardíaca de reciente instalación o empeoramiento de la existente) fueron más bajas en el grupo al que se colocó un VVIR-40 que en el grupo que recibió DDDR-70, con un porcentaje significativamente mayor de latidos ventriculares estimulados en el grupo DDDR-70 a los 3 meses de seguimiento (20).
Recientemente se ha descrito a las CVP frecuentes -con o sin episodios de taquicardia ventricular no sostenida- como una de las causas de miocardiopatía reversible (21). Se ha investigado mucho cuál sería el corte de la carga de CVP necesarias para desencadenar los mecanismos que llevan a las miocardiopatías. Se han propuesto diversos límites de corte de la carga de CVP que serviría para distinguir a los pacientes con MIA de aquellos que tienen una miocardiopatía dilatada primaria (22,23). Hay otras características -como por ejemplo el mayor tiempo de evolución de las CVP, presencia de TV no sostenida, CVP multiformes, y CVP del VD- que se pueden asociar con MIA (23). También se ha considerado a las taquiarritmias ventriculares idiopáticas sostenidas como causas comunes de MIT (24). En la figura 1 se resumen las causas de miocardiopatías inducidas por arritmia de origen ventricular.
Fisiopatología
La existencia de arritmia no necesariamente implica que tenga ésta que derivar en una MIT; todavía no se comprenden plenamente los mecanismos y vías responsables que la determinan en cada paciente individual. Entre los principales factores de riesgo que contribuyen a su aparición y al grado de disfunción miocárdica están el tipo de arritmia, la frecuencia cardíaca, la duración de la taquicardia, y la existencia de una cardiopatía de base. Otros factores incluyen edad, drogas administradas, y comorbilidades (6,7). Todos estos factores confluirían para determinar el momento de la aparición, la duración, y el grado de resolución de la MIT.
El modelo animal experimental de Whipple brinda información invalorable sobre los efectos que tienen los ritmos rápidos generados en aurículas o ventrículos sobre el miocardio, llegando a inducir disfunciones sistólicas y diastólicas severas.14 En otro modelo animal experimental, Zupan y col.25 demostraron que la disfunción ventricular comenzaba precozmente luego del inicio de la estimulación auricular o ventricular; la disfunción fue más pronunciada después de la estimulación ventricular, y se demostró que era reversible luego de cesar la estimulación. En humanos se han demostrado efectos similares de la estimulación a largo plazo (26).
En la figura 2 (7,25,27- 35) se resumen las alteraciones macroscópicas de la estructura y la función miocárdicas, las alteraciones neurohumorales y los cambios a nivel microscópico. De los posibles mecanismos primarios responsables de las modificaciones celulares y macroscópicas observadas, los que se proponen como predominantes son el agotamiento energético miocárdico y la isquemia miocárdica. En modelos de taquicardia persistente se ha demostrado el agotamiento de los depósitos de energía en forma de creatina, fosfocreatina, y trifosfato de adenosina, junto con reducción de los niveles de la actividad de Na-K-ATPasa, posibles consecuencias del incremento de la actividad de las enzimas del ciclo de Krebs, y lesión mitocondrial (36–39). Además, las alteraciones estructurales y funcionales de la red capilar miocárdica llevan a un deterioro de la reserva de flujo sanguíneo miocárdico e inducen isquemia miocárdica (32). Es posible que se produzca entonces una especie de hibernación miocárdica, que explicaría por qué esos cambios pueden revertirse parcial o completamente una vez que cesa la arritmia causal (2). Finalmente, el estrés oxidativo es otro de los mecanismos propuestos que contribuiría a la lesión miocárdica al producirse un desequilibrio entre las vías pro oxidantes y antioxidantes, dañando del ADN mitocondrial (40).

Asincronía
Las contracciones miocárdicas asincrónicas, por ejemplo por estímulos originados en el ventrículo derecho, o por bloqueo de rama, modifican la activación ventricular normal que pasa a través del sistema de His-Purkinje. Más allá de estar alterada la activación eléctrica, simultáneamente hay cambios de los patrones de activación mecánica que producen una redistribución de la carga y trabajo miocárdico con la consiguiente disminución de la efectividad de la contracción (41). Se utilizó centellografía con I123-meta-yodo-bencil-guanidina (MIBG) para estudiar la inervación adrenérgica del ventrículo izquierdo durante la estimulación crónica originada en la punta del ventrículo derecho. Se ha demostrado que la gran mayoría (89,7%) de los pacientes con marcapasos tenían defectos regionales de la captación de I123-MIBG, predominando en la pared inferior (92,3%) y apical (38,5%) (42). Luego de un período prolongado de estimulación apical del VD se pueden ver defectos de la perfusión miocárdica en hasta 2/3 de los pacientes (43). Las vías fisiopatológicas involucradas en la miocardiopatía inducida por CVP todavía no están claramente definidas, si bien estarían implicados mecanismos similares a los de la estimulación ventricular derecha. No se ha probado que la frecuencia cardíaca de hecho esté elevada; probablemente no difiera entre los pacientes con y sin miocardiopatía (44). El número de CVP -expresado como el porcentaje de CVP con respecto al número total de complejos QRS o como la suma de CVP por día- ha sido utilizado como criterio de miocardiopatía inducida por CVP, si bien no se han hecho ensayos que comparen las consecuencias fisiopatológicas entre los grupos de pacientes con diferentes cargas de CVP (22,44,45). Además, las CVP interpoladas —números totales y porcentajes de todas las CVP- han demostrado ser determinantes independientes de la miocardiopatía inducida por CVP (44).
La asincronía también resulta evidente en el caso de la FA. La pérdida de contracción auricular produce una reducción del 15%–20% del gasto cardíaco, y alteraciones de los tiempos de llenado y de las propiedades del VI, a causa de la asincronía aurículo-ventricular. La insuficiencia mitral empeora y se elevan las presiones enclavadas pulmonares (46). También se ha demostrado que la secuencia irregular de los intervalos RR produce consecuencias hemodinámicas adversas que son independientes de la frecuencia cardíaca(47). El mecanismo de Starling que relaciona la longitud de la miofibrilla con la intensidad de la contracción ventricular y la relación fuerza–intervalo son dos posibles mecanismos que vinculan la irregularidad de los intervalos RR y la reducción del gasto cardíaco.
La remodelación auricular como fenómeno resultante de la taquicardia incesante ha sido denominada MIT auricular (2). Los mecanismos que contribuyen a la remodelación del miocardio auricular –dilatación auricular y disfunción sistólica- difieren sustancialmente de los del miocardio ventricular. Las alteraciones del manejo del calcio que surgen de la regulación a la baja o la función alterada del canal de Ca(2+) Tipo L y un aumento de la extrusión de Ca (2+) vía el intercambiador de Na (+)/Ca (2+) han sido propuestos como los cambios iónicos primarios (48). Además, en un modelo animal de insuficiencia cardíaca congestiva inducida por una estimulación ventricular rápida experimental, se ha demostrado apoptosis tisular, infiltrado de células inflamatorias, y muerte celular. Los cambios máximos en la aurícula izquierda ocurrieron más precozmente y fueron mayores que en el ventrículo izquierdo, además de haber una activación más rápida de la proteinquinasa activada por el mitógeno y niveles elevados de factor de crecimiento y transformación -b1 en la aurícula izquierda (49).
Evolución y recuperación
Alteraciones hemodinámicas precoces se hacen evidentes casi inmediatamente de iniciadas las frecuencias ventriculares rápidas. En un modelo canino de insuficiencia cardíaca, la instalación aguda de la estimulación produjo una disminución de la presión arterial y del gasto cardíaco y aumentos de la presión enclavada pulmonar y de la aurícula derecha en 24 horas, persistiendo hasta luego de 3 semanas de la estimulación (50).
En la mayoría de los modelos animales, las modificaciones de las propiedades del llenado y de la contractilidad del VI ya se han completado a las 4–5 semanas de estimulación rápida (28,31). Además, se han analizado las curvas de presión–volumen utilizando registros intracardíacos antes e inmediatamente después de la restauración a corto plazo de la secuencia de activación ventricular normal en pacientes con marcapasos bicamerales crónicos (pasando de un DDD a un modo AAI). La elastancia telesistólica y su relación con la elastancia arterial efectiva mejoraron de forma aguda después de pasar a un modo de estimulación AAI (51).
Habitualmente los pacientes consultan cuando ya han pasado meses o años de insuficiencia cardíaca congestiva sintomática. Se considera que el tiempo que demora en recuperarse luego de instaurado el tratamiento depende de la frecuencia y de la duración de la arritmia. Durante la recuperación se ha demostrado una mejoría clínica rápida. Por el contrario, los volúmenes diastólicos del VI se mantienen aumentados en la ecocardiografía, mostrando una regresión gradual e incompleta a la normalización hacia las 4 semanas de la recuperación (52). Se espera que se produzca una recuperación casi completa de los síntomas y de la contractilidad del VI recién a los 3 meses de haberse logrado controlar el ritmo o la frecuencia cardíaca de la taquiarritmia.53
Se han publicado resultados similares en pacientes sometidos a ablación del nodo aurículo-ventricular y colocación de marcapasos debido a la persistencia de la FA con altas frecuencias ventriculares pese a un tratamiento médico óptimo.54 Por el contrario, Brignole y colaboradores en el reciente estudio aleatorizado AIRCRAFT no confirmaron tales diferencias significativas en la clase funcional de la Asociación del Corazón de Nueva York (NYHA) o en las medidas objetivas de la función cardíaca (55,56). Es probable que haya que considerar el posible efecto deletéreo de la estimulación apical del VD en este caso.
Hay otros dos aspectos que es preciso tener en cuenta. La hipertrofia del ventrículo izquierdo que persiste luego de semanas de haber interrumpido la estimulación puede ser atribuida a la respuesta post estimulación de los miocitos a los desencadenantes de hipertrofia. Esta respuesta puede aumentar aún más por la persistencia de la dilatación del VI (27). Lo que es más importante es que a pesar de normalizarse la FE, se ha demostrado que persiste la remodelación del VI (las dimensiones del VI y sus volúmenes quedan significativa y persistentemente elevados comparado con los controles) lo que tiene implicaciones en la duración y tipo de terapia con antiarrítmicos (57).
Diagnóstico y criterios propuestosA menudo es difícil hacer un diagnóstico definitivo de MIA. La mayoría de los pacientes tienen un cierto grado de disfunción sistólica o diastólica aurículoventricular; sin embargo, la arritmia causal puede no estar presente. Aun cuando se identifique una arritmia concomitantemente con la depresión de la función miocárdica, no siempre resulta factible establecer una relación causa–efecto. Por consiguiente, es necesario conservar un alto índice de sospecha. Dada la naturaleza retrospectiva del diagnóstico, la mayoría de los médicos acepta que una vez que existe una posibilidad teórica de miocardiopatía dilatada secundaria a arritmia, es fundamental aplicar esquemas de tratamiento antiarrítmico y esperar la resolución de los síntomas y la restauración parcial o completa de las propiedades estructurales y funcionales del miocardio. Fenelon y colaboradores han propuesto algunos criterios para el diagnóstico de MIT: 1) dilatación del corazón o insuficiencia cardíaca; 2) arritmia cardíaca crónica o muy frecuente (taquicardia supraventricular incesante, FA, o aleteo, taquicardia ventricular incesante). Además, sugieren que una taquicardia crónica que esté presente ³ 10 – 15% del día puede provocar una miocardiopatía y enfatizan que también debería sospecharse MIT en pacientes con coexistencia de arritmias y miocardiopatía (6).
Índices ecocardiográficos
Se han hecho intentos por predecir MIT aplicando índices ventriculares ecocardiográficos. Fujino y colaboradores (9) concluyeron que el tamaño de VI al ingreso era menor para los pacientes con MIT (diámetro telediastólico del VI: 57,6 ± 7,2 mm, diámetro telesistólico del VI: 49,4 ± 8,0 mm) que en el grupo con miocardiopatía dilatada (diámetro telediastólico del VI: 63,4 ± 8,8 mm, diámetro telesistólico de VI: 55,3 ± 9,6 mm, P <0,05) y que los pacientes con MIT tenían un mejor pronóstico (muerte cardíaca, hospitalización por insuficiencia cardíaca) durante el seguimiento. En otro estudio, un diámetro telediastólico del VI £ 61 mm predecía MIT con una sensibilidad de 100% y una especificidad de 71,4%. En pacientes con FE £ 30%, el diámetro telediastólico del VI £ 66 mm predecía MIT con una sensibilidad de 100% y una especificidad de 83,4%. Todos los pacientes con MIT mostraron mejorías de la FE ³ 15%, un hallazgo no demostrado en los pacientes con miocardiopatía no dilatada (delta-FE ³ 5%) (53). Se piensa que el menor agrandamiento del VI es provocado por ser un proceso relativamente más rápido, a diferencia del proceso degenerativo crónico que se observa en el contexto de miocardiopatías dilatadas primarias (58).
Nivel de asincronía
Todavía no han sido establecidos los criterios del nivel y tipo de asincronía necesarios para inducir una miocardiopatía como la que aparece durante la estimulación del VD. Se ha logrado demostrar que la disincronía del VI puede estar presente en ³ 50% de quienes requieren estimulación permanente del VD, y se asocia con deterioro de la función sistólica del VI y la clase funcional de NYHA (59). Sin embargo, todavía se está estudiando si la disincronía del VI que aparece justo después del inicio de la estimulación puede efectivamente provocar un deterioro funcional gradual del VI y la aparición de insuficiencia cardíaca congestiva (4).
Carga de los complejos ventriculares prematuros
En el contexto de las CVP como causa primaria de miocardiopatía, se ha demostrado que la disfunción del VI inducida por CVP no se limita a las CVP provenientes del ventrículo derecho (22,60). Además, una cierta carga de CVP puede facilitar el diagnóstico, al discriminar pacientes con miocardiopatía inducida de aquellos con arritmogénesis secundaria a la miocardiopatía. Diferentes autores han concluido en diferentes límites de corte de la carga de CVP. Baman y colaboradores propusieron una carga de CVP ³ 24% como asociada independientemente con una miocardiopatía inducida por CVP (sensibilidad 79%, especificidad 78%). Un punto de corte más bajo elevó la sensibilidad a 90%, pero redujo la especificidad a 58%. La carga de CVP más baja que producía miocardiopatía fue 10%. Otro criterio fue una FE anormal que mejorara por lo menos 15% o que se normalizara (³ 50%) después de un procedimiento de ablación efectivo (ver la siguiente sección de estrategias de tratamiento) (22). Hasdemir y colaboradores (24) propusieron otros predictores de miocardiopatía inducida por CVP, más allá de un límite de corte propuesto de ³ 16% (sensibilidad 100%, especificidad 87%). Se trata del género masculino, ausencia de síntomas, persistencia de CVP a lo largo del día, y presencia de CVP monomorfas repetitivas. Otro estudio reciente de Muñoz y colaboradores (23) concluye que una duración más prolongada de las CVP, presencia de taquicardia ventricular no sostenida, CVP multiformes, y CVP del VD pueden acompañarse de miocardiopatía, siendo el último hallazgo debido a la diferente carga de CVP necesaria para inducir miocardiopatía (carga ³ 10% para las CVP originadas en el VD contra =20% para CVP que se originaran en el VI). Finalmente se han hecho intentos por estratificar a los pacientes según el riesgo de miocardiopatía según el número total de CVP al día. Se concluyó que si los pacientes se dividían en tres grupos (<1.000/24 h, 1.000–10.000/24 h, y >10.000/24 h), la prevalencia de miocardiopatía fue 4%, 12%, y 34%, respectivamente (45).
Recientemente Buschan y Asirvatham han propuesto criterios en torno a la miocardiopatía inducida por CVP. Sugieren que individuos jóvenes por otra parte sanos, sin sustrato cardiovascular anormal, que tengan más de 20.000 CVP por día, no más de dos morfologías de CVP, CVP que se originaran de los tractos de salida o de los fascículos y con espesor de la pared miocárdica preservado (sin cicatrices demostradas por la ecocardiografía) son los mejores candidatos para presunción del diagnóstico de miocardiopatía inducida por CVP (61).
Estrategias de tratamiento
En la mayoría de los casos se puede establecer un diagnóstico definitivo sólo cuando la eliminación de la arritmia lleva a una mejora funcional y estructural. En otras palabras, las estrategias de tratamiento tienen que aplicarse suponiendo el diagnóstico, una hipótesis que puede confirmarse retrospectivamente. Hay diferentes tipos de estrategias terapéuticas adecuadas para diferentes arritmias, dado que la meta primaria del tratamiento difiere, apuntando a la frecuencia cardíaca/ritmo o alivio de la asincronía ventricular. Nuestra estrategia terapéutica consiste en regímenes farmacológicos, intervenciones con catéter y dispositivos.
Tratamientos médicos
Casi todas las categorías de agentes antiarrítmicos han sido utilizados en casos de miocardiopatías inducidas por arritmia. Los beta bloqueantes, digital, inhibidores de los canales de calcio no di-hidropiridina, o combinaciones son medicamentos utilizados ampliamente para controlar la frecuencia en la FA (62). Se considera que la estrategia de control de la frecuencia es no inferior al control del ritmo, siempre y cuando la frecuencia cardíaca no se mantenga sin controlar, excepto en pacientes que presentan sintomatología severa (63,64). Si no se puede lograr el control de la frecuencia ventricular a largo plazo deberían aplicarse estrategias de control del ritmo, lo que incluye la cardioversión con corriente directa y agentes Clase Ic y III de Vaughan Williams (ver también terapias con catéter) (62). Hay una serie de arritmias supraventriculares también se pueden tratar, por lo menos inicialmente, con agentes antiarrítmicos, si bien habitualmente en casos de miocardiopatía, es posible que el tratamiento de elección sea la ablación (53).
Cuando se considere que las CVP frecuentes sean la causa primaria, se puede recurrir a dos estrategias alternativas de tratamiento. La primera sería la administración de un agente antiarrítmico, fundamentalmente amiodarona o, en el caso de las CVP que se originan en los tractos de salida ventriculares, se puede administrar inhibidores de los canales de calcio no di-hidropiridina. Estos últimos pueden ser utilizados siempre y cuando no produzcan un deterioro del estado clínico del paciente debido a sus efectos inotrópicos negativos. La amiodarona debería administrarse solo de manera transitoria, ya que sus efectos tóxicos pueden aparecer inesperadamente. Su uso ha sido propuesto para 3–6 meses en pacientes cuando no hay certidumbre con respecto al diagnóstico final, seguido de la ablación focal de las CVP como tratamiento permanente si se reducen las CVP y se recupera la función cardíaca (61).
Sin embargo, debemos tener en cuenta que aun cuando los agentes antiarrítmicos sean capaces de controlar el mecanismo causante de la miocardiopatía, es posible que la remodelación miocárdica inducida sea resistente al tratamiento. Tal como se describió en los párrafos anteriores, Dandamudi y colaboradores (57) demostraron la persistencia de la dilatación del VI después del tratamiento de la MIT. Para el manejo de estos volúmenes ventriculares aumentados se ha propuesto la administración a largo plazo de beta-bloqueantes e inhibidores de la enzima convertidora de angiotensina (2,57,65).
Terapias con catéter
Hay muchos estudios que ponen de manifiesto el papel dominante de las intervenciones con catéter en el tratamiento de las arritmias que provocan miocardiopatía. Es importante destacar que su rango en el algoritmo terapéutico difiere según el tipo específico de arritmia, el grado de disfunción miocárdica, y la severidad de los síntomas. La ablación logra abolir la arritmia culpable mucho más frecuentemente que el tratamiento antiarrítmico; además, evita el uso de agentes farmacológicos, que entre otros problemas, plantean el peligro de efectos proarrítmicos.
Los procedimientos ablativos son aplicables para casi cualquier tipo de arritmia supraventricular, como ser la taquicardia auricular, el aleteo auricular, las taquicardias de reentrada nodal aurículo-ventricular, o de reentrada aurículo-ventricular, con excelentes resultados en cuanto al alivio de los síntomas y la regresión de la disfunción miocárdica (10–13). Como implicación clínica importante del éxito de la ablación, Pizzale y colaboradores (10) relatan que los pacientes que cumplen con los criterios para la colocación de un cardiodesfibrilador implantable (CDI) antes de la ablación, pueden tener una mejora tal de su función ventricular que dejan de ser candidatos para el CDI después de la ablación.
Se considera que las estrategias de ablación en la FA son más complejas. Si hay una mala respuesta al control del ritmo o la frecuencia a pesar de la medicación, o si falla la ablación de la aurícula izquierda, se puede realizar ablación del nodo aurículo-ventricular para mejorar los síntomas y el pronóstico (62). Sin embargo, se sigue discutiendo si la fracción de eyección se puede recuperar o si se deteriora a largo plazo debido a la estimulación permanente del VD (54–56). La ablación de la aurícula izquierda es un procedimiento bien documentado para la eliminación de las FA paroxísticas y persistentes, cuando fracasan los intentos con los antiarrítmicos (62,66). Varios autores han abordado el tema de la ablación de la FA en pacientes con depresión de la función del VI (67,68). Gentlesk y colaboradores comunicaron una mejoría de la FE de por lo menos 5% en 82% de ellos, mientras que la FE se normalizó en 72% luego de la ablación. Además, la tasa de éxitos parece similar entre los grupos de pacientes con función ventricular normal y deprimida (68). Por otro lado, mientras que Lutomsky y colaboradores (69) confirmaron la mejoría de la FE usando resonancia magnética cardíaca, también relatan una reducción significativa de la tasa de éxitos del procedimiento de ablación en pacientes con función ventricular deprimida, planteando un mecanismo multifactorial como responsable de la disfunción miocárdica de base.
Los sitios de origen únicos o múltiples de CVP también se pueden detectar y eliminar mediante ablación con radiofrecuencia luego de un mapeo de activación y/o topoestimulación, cuando fallan los intentos con agentes antiarrítmicos (22–24,60). Los criterios de éxito difieren. Baman y colaboradores (22) consideraron el procedimiento de ablación eficaz si se lograba abolir por lo menos 80% de la carga de CVP, mientras que Muñoz y colaboradores (23) han establecido criterios más estrictos de eliminación completa e incapacidad de inducción de la arritmia clínica postablación. Se necesita un período de por lo menos 3–6 meses después de la ablación para acceder al estado clínico, mejora de la función miocárdica (si está presente) y detección temprana de recurrencia.
Terapia de resincronización
Los sistemas modernos de estimulación bicamerales garantizan la sincronización aurículo-ventricular e incorporan algoritmos sofisticados que minimizan la estimulación ventricular, si no es inevitable. Actualmente hay mucha investigación sobre cómo evitar los efectos deletéreos de la estimulación crónica del VD. Se han propuesto diferentes sitios de estimulación, tales como el tracto de salida del VD, el tabique del VD y la región de His (70–72).
En la práctica clínica, sin embargo, lo que ha sido más ampliamente propuesto como alternativa más confiable es la estimulación biventricular. En pacientes con FE normal e indicación convencional de implantación de marcapasos, la estimulación biventricular puede evitar los efectos deletéreos de la estimulación del VD en la función miocárdica (73). La estimulación biventricular en pacientes con disfunción ventricular y una indicación para marcapasos permanente (estudio HOBIPACE) también redujo los volúmenes telediastólicos y telesistólicos del VI comparado con pacientes que recibían estimulación convencional del VD. La fracción de eyección del ventrículo izquierdo fue más elevada en quienes recibieron marcapasos biventriculares (74). Además, recientemente se ha comunicado que los pacientes que inicialmente se estimulaban desde el VD y desarrollaban miocardiopatía atribuida a la estimulación, cuando eran sometidos a estimulación biventricular lograban restaurar parcialmente la función del VI, los volúmenes ventriculares y regresión parcial de los síntomas.75 Un “upgrade” alivia los síntomas, así como la tolerancia al ejercicio (75,76). Los datos han confirmado el efecto de la estimulación biventricular en dichos pacientes en términos del alivio de la disincronía ventricular (76,77).
Finalmente se han comunicado casos de restauración completa de la función sistólica y del volumen miocárdico mediante estimulación biventricular en pacientes con bloqueo de rama izquierda, y miocardiopatía dilatada no isquémica, destacando la posible relación causa–efecto entre los trastornos de la conducción intraventricular y la disfunción miocárdica (78).
Seguimiento y riesgo de recurrenciaLa duración óptima del seguimiento no ha sido definida, si bien la mayoría de los pacientes parecen recuperarse dentro de los 3–6 meses (22,53). Pueden ser necesarios estudios y registros ecocardiográficos para determinar la recurrencia de la arritmia. Si bien un cierto grado de recuperación es lo habitual, una forma de miocardiopatía irreversible no debería llevar a la exclusión de diagnóstico de MIT.
En caso de recurrencia, se ha comunicado deterioro de la función miocárdica y síntomas severos, siendo la velocidad del deterioro significativamente más rápida que en el período inicial. Además, si ocurre una recurrencia, se ha descrito una relación con la muerte súbita cardíaca, pero no se comprenden plenamente las vías fisiopatológicas posibles (79).
¿Existe una respuesta al enigma?
Las formas reversibles de miocardiopatías debidas a causas arrítmicas representan un porcentaje apreciable de los diagnósticos en pacientes que se presentan con disfunción miocárdica y síntomas de insuficiencia cardíaca congestiva (80). Sin embargo, aun si la arritmia causal es evidente, la aclaración de la relación causa-efecto algunas veces representa un “dolor de cabeza” diagnóstico para el médico tratante, dado que el diagnóstico puede confirmarse sólo retrospectivamente. Los criterios propuestos hasta la fecha varían sustancialmente entre los investigadores, especialmente en el contexto de las CVP frecuentes. De hecho, se necesitan registros de mayor tamaño y ensayos aleatorizados para diagnosticar y estratificar a los pacientes de una manera más racional, según los parámetros ecocardiográficos y la carga de la arritmia. Además, habría que buscar las variaciones genéticas que predisponen a los pacientes a esas formas de miocardiopatía. En conclusión, hay que estudiar minuciosamente a cada paciente individual, y tratarlo de manera competente, ya que nunca debería darse una respuesta automática a dicho dilema.
Bibliografía
1. Gallagher JJ. Tachycardia and cardiomyopathy: the chicken-egg dilemma revisited. J Am Coll Cardiol 1985;6:1172–3.
2. Khasnis A, Jongnarangsin K, Abela G, Veerareddy S, Reddy V, Thakur R. Tachycardia-induced cardiomyopathy: a review of literature. Pacing Clin Electrophysiol 2005;28:710-21.
3. Chugh SS, Shen WK, Luria DM, Smith H. First evidence of premature ventricular complex-induced cardiomyopathy: a potentially reversible cause of heart failure. J Cardiovasc Electrophysiol 2000;11:328-9.
4. Tops LF, Schalij MJ, Bax JJ. The effects of right ventricular apical pacing on ventricular function and dyssynchrony: implications for therapy. J Am Coll Cardiol 2009;54:764-76.
5. Redfield MM, Kay GN, Jenkins LS, MIanulli M, Jensen DN, Ellenbogen KA. Tachycardia-related cardiomyopathy: a common cause of ventricular dysfunction in patients with atrial fibrillation referred for atrioventricular ablation. Mayo Clin Proc 2000;75:790-5.
6. Fenelon G, Wijins W, Andries E, Brugada P. Tachycardiomyopathy: mechanisms and clinical implications. Pacing Clin Electrophysiol 1996;19:95- 106.
7. Shinbane JD, Wood MA, Jensen DN, Ellenbogen KA, Fitzpatrick AP, Sheinman MM. Tachycardia-induced cardiomyopathy: a review of animal models and clinical studies. J Am Coll Cardiol 1997;29:709-15.
8. Gossage AM, Braxton Hicks JA. On auricular fibrillation. Q J Med 1913;6:435-40.
9. Fujino T, Yamashita T, Suzuki S, Sugiyama H, Sagara K, Sawada H et al. Characteristics of congestive heart failure accompanied by atrial fibrillation with special ref-erence to tachycardia-induced cardiomyopathy. Circ J 2007;71:936-40.
10. Pizzale S, Lemery R, Green MS, Gollob MH, Tang AS, Birnie DH. Frequency and predictors of tachycardia-induced cardiomyopathy in patients with persistent atrial flutter. Can J Cardiol 2009;25:469-72.
11. Furushima H, Chinushi M, Sugiura H, Aizawa Y. Radiofrequency catheter ablation for incessant atrioventricular nodal reentrant tachycardia normalized H-V block associated with tachycardia-induced cardiomyopathy. J Electrocardiol 2004;37: 315-9.
12. Medi C, Kalman JM, Haqqani H, Vohra JK, Morton JB, Sparks PB et al. Tachycardia- mediated cardiomyopathy secondary to focal atrial tachycardia. J Am Coll Cardiol 2009; 53:1791-7.
13. Bensler JM, Frank CM, Rasavi M, Rasekh A, Saeed M, Haas PC, et al. Tachycardia- mediated cardiomyopathy and the permanent form of junctional reciprocating tachycardia. Texas Heart Inst J 2010;37:695-8.
14. Whipple GH, Sheffield LT, Woodman EG, Theophilis C, Friedman S. Reversible congestive heart failure due to chronic rapid stimulation of the normal heart. Proc N Engl Cardiovasc Soc 1962;20:39-40.
15. Anter E, Jessup M, Callans DJ. Atrial fibrillation and heart failure. Treatment considerations of a dual epidemic. Circulation 2009;119:2516-25.
16. Edner M, Caidahl K, Bergfeldt L, Darpo¨ B, Edvardsson N, Rosenqvist M. Prospective study of left ventricular function after radiofrequency ablation of atrioventricu- lar junction in patients with atrial fibrillation. Br Heart J 1995;74:261-7.
17. Prinzen FW, Peschar M. Relation between the pacing inducedsequence of activa- tion and left ventricular pump function in animals. Pacing Clin Electrophysiol 2002; 25:484-98.
18. Tops LF, Schalij MJ, Holman ER, van Erven L, van der Wall EE, Bax JJ. Right ventricular pacing can induce ventricular dyssynchrony in patients with atrial fibrillation after atrioventricular node ablation. J Am Coll Cardiol 2006;48:1642-8.
19. Sweeney MO, Hellkamp AS, Ellenbogen KA, Greenspon AJ, Freedman RA, Lee KL et al. Adverse effect of ventricular pacing on heart failure and atrial fibrillation among patients with normal baseline QRS duration in a clinical trial of pacemaker therapy for sinus node dysfunction. Circulation 2003;107:2932-7.
20. Wilkoff BL, Cook JR, Epstein AE, Greene HL, Hallstrom AP, Hsia H et al. On behalf of the Dual Chamber and VVI Implantable Defibrillator Trial Investigators. Dual chamber pacing or ventricular backup pacing in patients with an implantable defibrillator: the Dual Chamber and VVI Implantable Defibrillator (DAVID) trial. JAMA 2002;288: 3115-23.
21. Duffee DF, Shen WK, Smith HC. Suppression of frequent prematureventricular contractions and improvement of left ventricular function in patients with pre- sumed idiopathic dilated cardiomyopathy. Mayo Clin Proc 1998;73:430-3.
22. Baman TS, Lange DC, Ilg KJ, Gupta SK, Liu T, Alguire C et al. Relationship between burden of premature ventricular complexes and left ventricular function. Heart Rhythm 2010;7:865-9.
23. Del Caprio Munoz F, Syed FF, Noheria A, Cha Y, Friedman PA, Hammill SC et al. Characteristics of premature ventricular complexes as correlates of reduced left ventricular systolic function: study of the burden, duration, coupling interval, morphology and site of origin of PVCs. J Cardiovasc Electrophysiol 2011;22:79-8.
24. Hasdemir C, Ulucan C, Yavuzgil O, Yuksel A, Kartal Y, Simsek E et al. Tachycardia-induced cardiomyopathy in patients with idiopathic ventricular arrhythmias: the incidence, clinical and electrophysiological characteristics, and the predictors. J Cardiovasc Electrophysiol 2011;22:663-8.
25. Zupan I, Rakovec P, Budihna N, Brecelj A, Kozelj M. Tachycardia-induced cardio- myopathy in dogs; relation between chronic supraventricular and chronic ven- tricular tachycardias. Int J Cardiol 1996;56:75-81.
26. Thackray SD, Witte KK, Nikitin NP, Clark AL, Kaye GC, Cleland JG. The preva- lence of heart failure and asymptomatic left ventricular systolic dysfunction in a typical regional pacemaker population. Eur Heart J 2003;24:1143-52.
27. Tomita M, Spinale FG, Crawford FA, Zile MR. Changes in left ventricular volume, mass and function during the development and regression of supraventricular tachycardia-induced cardiomyopathy. Disparity between recovery of systolic versus diastolic function. Circulation 1991; 83:635-44.
28. Ohno M, Cheng CP, Little WC. Mechanism of altered patterns of left ventricular filling during the development of congestive heart failure. Circulation 1994;89:2241-50.
29. Byrne MJ, Raman JS, Alferness CA, Esler MD, Kaye DM, Power JM. An ovine model of tachycardia-induced degenerative dilated cardiomyopathy and heart failure with prolonged onset. J Card Fail 2002;8:108-15.
30. Massoure PL, Jais P, Sacher F, Haissaguerre M. Tachycardia-induced right ventricu- lar cardiomyopathy: epicardial radiofrequency ablation of an unusual and unexpected ventricular tachycardia. J Cardiovasc Electrophysiol 2009; 20:813-7.
31. Timek TA, Dagum P, Lai DT, Liang D, Daughters GT, Ingels NB et al. Pathogenesis of mitral regurgitation in tachycardia-induced cardiomyopathy. Circulation 2001; 104(Suppl I):I-47-53.
32. Spinale FG, Tanaka R, Crawford FA, Zile MR. Changes in myocardial blood flow during development of and recovery from tachycardia-induced cardiomyopathy. Circulation 1992;85:717-29.
33. Moe GW, Stopps TP, Angus C, Forster C, DeBold AJ, Armstrong PW. Alterations in serum sodium in relation to atrial natriuretic factor and other neuroendocrine variables in experimental pacing-induced heart failure. J Am Coll Cardiol 1989;13: 173-9.
34. Zellner JL, Spinale FG, Eble DM, Hewett KW, Crawford FA Jr. Alterations in myocyte shape and basement membrane attachment with tachycardia-induced heart failure. Circ Res 1991;69:590-600.
35. Tanaka R, Fulbright BM, Mukherjee R, Burchell SA, Crawford FA, Zile MR et al. The cellular basis for the blunted response to beta-adrenergic stimulation in supraventricular tachycardia-induced cardiomyopathy. J Mol Cell Cardiol 1993;25: 1212-33.
36. Moe GW, Montgomery C, Howard RJ, Grima EA, Armstrong PW. Left ventricular myocardial blood flow, metabolism and effects of treatment with enalapril: further insights into the mechanisms of canine experimental pacing-induced heart failure. J Lab Clin Med 1993;131:294-301.
37. Spinale FG, Clayton C, Tanaka R, Fulbright BM, Mukherjee R, Schulte BA et al. Myocardial Na(+), K(+)-ATPase in tachycardia induced cardiomyopathy. J Mol Cell Cardiol 1992;24:277-94.
38. Coleman HN 3rd, Taylor RR, Pool PE, Whipple GH, Covell JW, Ross J Jr et al. Congestive heart failure due to chronic rapid stimulation of the normal heart.Am Heart J 1971;81:790-8.
39. O’Brien PJ, Ianuzzo CD, Moe GW, Stopps TP, Armstrong PW. Rapid ventricular pacing of dogs to heart failure: biochemical and physiological studies. Can J Physiol Pharmacol 1990;68:34-9.
40. Saavedra WF, Paolocci N, St John ME, Skaf MW, Stewart GC, Xie JS et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res 2002;90: 297-304.
41. Prinzen FW, Hunter WC, Wyman BT, McVeigh ER. Mapping o fregional myocardial strain and work during ventricular pacing: experimental study using magnetic resonance imaging tagging. J Am Coll Cardiol 1999; 33:1735-42.
42. Simantirakis EN, Prassopoulos VK, Chrysostomakis SI, Kochiadakis GE, Koukouraki SI, Lekakis JP et al. Effects of asynchronous ventricular activation on myocardial adrenergic innervations in patients with permanent dual-chamber pacemakers. Eur Heart J 2001;22:323-32.
43. Skalidis EI, Kochiadakis GE, Koukouraki SI, Chrysostomakis SI, Igoumenidis NI, Karkavitsas NS et al. Myocardial perfusion in patients with permanent ventricular pacing and normal coronary arteries. J Am Coll Cardiol 2001;37:124-9.
44. Olgun H, Yokokawa M, Baman T, Kim HM, Armstrong W, Good E et al. The role of interpolation in PVC-induced cardiomyopathy. Heart Rhythm 2011;8:1046-49.
45. Kanei Y, Friedman M, Ogawa N, Hanon S, Lam P, Schweitzer P. Frequent premature ventricular complexes originating from the right ventricular outflow tract are associated with left ventricular dysfunction. Ann Noninvasive Electrocardiol 2008;13: 81-5.
46. Clark DM, Plumb VJ, Kay GN. The hemodynamics of atrial fibrillation: the inde- pendent effect of an irregular RR interval. Circulation 1995;92(Suppl I):1-141.
47. Clark DM, Plumb VJ, Epstein AE, Kay GN. Hemodynamic effects of an irregular sequence of ventricular cycle lengths during atrial fibrillation. J Am Coll Cardiol 1997;30:1039-45.
48. Schotten U, Greiser M, Benke D, Buerkel K, Ehrenteidt B, Stellbrink C et al. Atrial fibrillation-induced atrial contractile dysfunction: a tachycardiomyopathy of a dif- ferent sort. Cardiovasc Res 2002;53:192-201.
49. Hanna N, Cardin s, Leung TK, Nattel S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc Res 2004;63:236-44.
50. Wilson RR, Douglas P, Hickey WF, Lanoce V, Ferrano N, Muhammad A et al. Experimental congestive heart failure produced by rapid ventricular pacing in the dog: cardiac effects. Circulation 1987;75:857-67.
51. Simantirakis EN, Kochiadakis GE, Vardakis KE, Igoumenidis NE, Chrysostomakis SI, Vardas PE. Left ventricular mechanics and myocardial blood flow following restoration of normal activation sequence in paced patients with long-term right ventricular apical stimulation. CHEST 2003;124:233-41.
52. Howard RJ, Stopps TP, Moe GW, Gotlieb A, Armstrong PW. Recovery from heart failure: structural and functional analysis in a canine model. Can J Physiol Phar- macol 1988;66:1505-12.
53. Jeong YH, Choi KJ, Song JM, Hwang ES, Park KM, Nam GB. Diagnostic approach and treatment strategy in tachycardia-induced cardiomyopathy. Clin Cardiol 2008; 31:172-8.
54. Kay GN, Ellenbogen KA, Giudici M, Redfield MM, Jenkins LS, Mianulli M et al. The Ablate and Pace Trial: a prospective study of catheter ablation of the AV conduc- tion system and permanent pacemaker implantation for treatment of atrial fibril- lation. APT Investigators. J Interv Card Electrophysiol 1998;2:121-35.
55. Brignole M, Menozzi C, Gianfranchi L, Musso G, Mureddu R, Bottoni N et al. Assessment of atrioventricular junction ablation and VVIR pacemaker versus pharmacological treatment in patients with heart failure and chronic atrial fibril- lation: a randomized, controlled study. Circulation 1998;98:953-60.
56. Weerasooriya R, Davis M, Powell A, Szili-Torok T, Shah C, Whalley D et al. The Australian intervention randomized control of rate in atrial fibrillation trial (AIR- CRAFT). J Am Coll Cardiol 2003;41:1697-702.
57. Dandamudi G, Rampurwala AY, Mahenthiran J, Miller JM, Das MK. Pesistent left ventricular dilatation in tachycardia-induced cardiomyopathy patients after appropriate treatment and normalization of ejection fraction. Heart Rhythm 2008;5:1111-4.
58. Lishmanov A, Chockalingam P, Senthilkumar A, Chockalingam A. Tachycardia-induced cardiomyopathy: evaluation and therapeutic options. Congest Heart Fail 2010;16:122-6.
59. Tops LF, Suffoletto MS, Bleeker GB, Boersma E, van der Wall EE, Gorcsan J III et al. Speckle-tracking radial strain reveals left ventricular dyssynchrony in patients with permanent right ventricular pacing. J Am Coll Cardiol 2007;50:1180-8.
60. Bogun F, Crawford T, Reich S, Koelling TM, Armstrong W, Good E et al. Radio- frequency ablation of frequent, idiopathic premature ventricular complexes: comparison with a control group without intervention. Heart Rhythm 2007;4:863-7.
61. Bhushan M, Asirvatham SJ. The conundrum of ventricular arrhythmia and cardio- myopathy: which abnormality came first? Curr Heart Fail Rep 2009;6:7-13.
62. European Heart Rhythm Association; European Association for Cardio-Thoracic Surgery, Camm AJ, Kirchhof P, Lip GY, Schotten U, Savelieva I, Ernst S et al. Guidelines for the management of atrial fibrillation: the Task Force for the Man- agement of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Europace 2010;12:1360-420.
63. Wyse DG, Waldo AL, DiMarco JP, Domanski MJ, Rosenberg Y, Schron EB et al. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002;347:1825-33.
64. Roy D, Talajic M, Nattel S, Wyse DG, Dorian P, Lee KL et al. Rhythm control versus rate control for atrial fibrillationand heart failure. N Engl J Med 2008; 358:2667-77.
65. Ramesh Iyer V. Ventricular dysfunction: tachycardia induced cardiomyopathy Indian Pacing Electrophysiol J 2008;8:202-10.
66. Nair GM, Nery PB, Diwakaramenon S, Healey JS, Connolly SJ, Morillo CA. A systematic review of randomized trials comparing radiofrequency ablation with anti-arrhythmic medications in patients with atrial fibrillation. J Cardiovasc Electrophysiol2009;20:138-44.
67. Efremidis M, Sideris A, Xydonas S, Letsas KP, Alexanian IP, Manolatos D et al. Ablation of atrial fibrillation in patients with heart failure: reversal of atrial and ven- tricular remodeling. Hellenic J Cardiol 2007;48:19-25.
68. Gentlesk PJ, Sauer WH, Gerstenfeld EP, Lin D, Dixit S, Zado E et al. Reversal of left ventricular dysfunction following ablation of atrial fibrillation. J Cardiovasc Elec- trophysiol 2007;18:9-14.
69. Lutomsky BA, Rostock T, Koops A, Steven D, Mullerleile K, Servatius H et al. Catheter ablation of paroxysmal trial fibrillation improves cardiac function: a prospective study on the impact of atrial fibrillation ablation on left ventricular function assessed by magnetic resonance imaging. Europace 2008;10:593-9.
70. Victor F, Leclercq C, Mabo P, Pavin D, Deviller A, de Place C et al. Optimal right ventricular pacing site in chronically implanted patients: a prospective randomized crossover comparison of apical and outflow tract pacing. J Am Coll Cardiol 1999; 33:311-6.
71. Bourke JP, Hawkins T, Keavey P, Tynan M, Jamieson S, Behulova R et al. Evolution of ventricular function during permanent pacing from either right ventricular apex or outflow tract following AV-junctional ablation for atrial fibrillation. Europace 2002;4:219-28.
72. Deshmukh PM, Romanyshyn M. Direct His-bundle pacing: present and future. Pacing Clin Electrophysiol 2004;27:862-70.
73. Yu CM, Chan JY, Zhang Q, Omar R, Yip GW, Hussin A et al. Biventricular pacing in patients with bradycardia and normal ejection fraction. N Eng J Med 2009;361:2123-34.
74. Kindermann M, Hennen B, Jung J, Geisel J, Bohm M, Frohlig G. Biventricular versus conventional right ventricular stimulation for patients with standard pacing indica- tion and left ventricular dysfunction: the Homburg Biventricular Pacing Evaluation (HOBIPACE). J Am Coll Cardiol 2006;47:1927-37.
75. Nazeri A, Massumi A, Rasekh A, Saeed M, Frank C, Razavi M. Cardiac resynchro- nization therapy in patients with right ventricular pacing-induced cardiomyopathy. Pacing Clin Electrophysiol 2010;33:37-40.
76. Leclercq C, Cazeau S, Lellouche D, Fossati F, Anselme F, Davy JM et al. Upgrading- from single chamber right ventricular to biventricular pacing in permanently paced patients with worsening heart failure: The RD-CHF Study. Pacing Clin Electrophysiol 2007;30:23-30.
77. Laurenzi F, Achilli A, Avella A, Peraldo C, Orazi S, Perego GB et al. Biventricular- upgrading in patients with conventional pacing system and congestive heartfailure: results and response predictors. Pacing Clin Electrophysiol 2007;30:1096-104.
78. Fujii B, Takami M. Normalization of left ventricular function following cardiac resynchronization therapy—left bundle branch block as a potential etiology of dilated cardiomyopathy. Circ J 2008;72:1030-3.
79. Nerheim P, Birger-Botkin S, Lubna Piracha DO, Olshansky B. Heart failure and sudden cardiac death in patients with tachycardia-induced cardiomyopathy and recurrent tachycardia. Circulation 2004;110:247-52.
80. Huffman C, Wagman G, Fudim M, Zolty R, Vittorio T. Revesible cardiomyop- athies—a review. Transplant Proc 2010;42:3673-8.

