










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Curriculum ScienTI
Curriculum ScienTIRelated links
Share

 Permalink
PermalinkRevista Uruguaya de Cardiología
On-line version ISSN 1688-0420
Rev.Urug.Cardiol. vol.27 no.2 Montevideo Aug. 2012
Consenso
Consenso de prevención primaria y secundaria de muerte súbita
Sociedad Argentina de Cardiología. Sociedad Uruguaya de Cardiología
Consenso de prevención primaria y secundaria de muerte súbita
Sociedad Argentina de Cardiología. Sociedad Uruguaya de Cardiología.
(Con colaboración del Consejo Argentino de Residentes de Cardiología)
Director
Dr. Enrique Oscar Retyk (MTSAC)
Coordinación General
Dr. Andrés Bochoeyer
Por Área de Normatizaciones y Consensos
Dr. Eduardo Alberto Sampó (MTSAC)
Coordinador por la SUC
Dr. Alejandro Cuesta
Coordinadores
Dr. Mauricio Abello (MTSAC)
Dr. César Cáceres Monié
Dr. Claudio De Zuloaga (MTSAC)
Dr. José Gant López
Dr. José Luis González (MTSAC)
Dr. Carlos Labadet (MTSAC)
Dr. Gustavo Maid
Dr. Claudio Militello (MTSAC)
Dr. Alberto Sciegata (MTSAC)
Comité de Redacción
Dr. Rafael Salvador Acunzo (MTSAC)
Dra. Karina Alonso
Dr. Daniel Aguerre Banina (SUC)
Dr. César Belziti (MTSAC)
Dr. Carlos Boissonnet (MTSAC)
Dr. Guillermo Bortman (MTSAC)
Dr. Martín Nicolás Calvelo (CONAREC)
Dr. Horacio Casabé (MTSAC)
Dr. Pedro Chiesa (SUC)
Dr. Alejandro Cueto
Dr. Felipe Deketele
Dr. Darío Di Toro (MTSAC)
Dr. Adrián Fernández
Dr. Pablo Fernández (SUC)
Dr. Alejandro Franco (MTSAC)
Dr. Diego Colla Freire
Dr. Juan Fuselli (MTSAC)
Dr. Juan Gagliardi (MTSAC)
Dr. Néstor Galizio (MTSAC)
Dr. Sebastián Gallino
Dr. Nicolás González (CONAREC)
Dr. Javier Guetta
Dra. Marianna Guerchicoff
Dr. Claudio Hadid
Dra. Isabel Victoria Konopka (MTSAC)
Dra. María Victoria Lafuente
Dr. Rubén Laiño (MTSAC)
Dra. Mariela Lujambio (SUC)
Dra. Florencia Meiller
Dr. José Moltedo (MTSAC)
Dr. Gerardo Nau (MTSAC)
Dr. Pablo Pieroni (CONAREC)
Dr. Horacio Quiroga Ponce
Dr. Walter Reyes (SUC)
Dr. Álvaro Rivara (SUC)
Dr. Carlos Rivas
Dr. Rodolfo Sansalone
Dr. Iván Tello Santacruz
Dra. Natalia Schnetzer
Dra. Andrea Simeone (SUC)
Dra. Amelia Stefani
Dra. Palmira Vanzini (SUC)
Dr. Gonzalo Varela (SUC)
Comité de Revisión
Dr. Sergio Dubner (MTSAC)
Dr. José Estepo (MTSAC)
Dr. Enrique Fairman (MTSAC)
Dr. Hugo Grancelli (MTSAC)
Dr. Alberto Giniger (MTSAC)
Dr. Rubén Laiño (MTSAC)
Dr. Oscar Oseroff (MTSAC)
Dr. Rafael Rabinovich (MTSAC)
Introducción
La prevención de la muerte súbita (MS) representa un gran desafío para la comunidad médica en general y hace que pese una gran responsabilidad sobre los especialistas en la materia. Siguiendo este espíritu de compromiso es que a través de las Sociedades Científicas hemos creado un grupo de trabajo para discutir y establecer consensos sobre los aspectos más relevantes del tema.
Se define como MS a la muerte natural, de causa cardiovascular, que se produce en forma inesperada, con un corto intervalo desde el inicio de los síntomas desencadenantes, habitualmente menor de una hora o que acontece durante el sueño.
La MS es un problema de considerable magnitud, ya que representa la mitad de las muertes cardiovasculares y 25% del total de las muertes en adultos. Aproximadamente la mitad de las veces puede ocurrir en personas sin enfermedad cardíaca conocida, como expresión de un primer episodio.
Si bien en valores absolutos la mayoría de las MS se producen en individuos sanos, su incidencia en la población general es baja y se incrementa a medida que se seleccionan subpoblaciones más graves. Estos pacientes con factores de riesgo elevado de MS representan la minoría en términos epidemiológicos. De este análisis se desprenden dos escenarios posibles:
1. En la población sin claros marcadores de riesgo de MS, el núcleo del tratamiento es el control de los factores de riesgo coronario, la consulta temprana ante la sospecha de síntomas de cardiopatía, la capacitación en la asistencia del paro cardiorrespiratorio y la disponibilidad de desfibriladores automáticos en sitios de alta concentración de personas.
2. En los subgrupos de mayor riesgo, la prevención justifica la adopción de medidas activas y eventualmente costosas para prevenir la MS. En consecuencia, dichas terapéuticas solo se aplican a un pequeño porcentaje de la población general que la presentará.
La enfermedad cardíaca estructural y/o inflamatoria constituye el principal sustrato en la fisiopatología de la MS. Sin embargo, alrededor de 5% de los episodios de MS ocurren en personas sin cardiopatía demostrable, particularmente en la población más joven. Existe una gran variedad de anormalidades electrofisiológicas “primarias” que contribuyen a la MS en pacientes sin cardiopatía estructural. La importancia de reconocer estos casos radica en que si se logra impedir que una eventual recurrencia arrítmica derive en paro cardíaco y muerte repentina, el pronóstico a largo plazo es excelente debido a la ausencia de cardiopatía estructural.
Este Consenso es fruto del esfuerzo conjunto del Área de Normatizaciones y Consensos, del Consejo de Electrofisiología de la Sociedad Argentina de Cardiología y de un grupo destacado de expertos de la Sociedad Uruguaya de Cardiología. Además de los capítulos de prevención de MS en las distintas miocardiopatías (hipertrófica, idiopática, isquémica), hemos desarrollado otros temas específicos de la especialidad, como síndrome de QT largo, síndrome de Brugada y displasia arritmogénica del ventrículo derecho (VD). También nos propusimos plantear recomendaciones en un tema de exclusivo interés, ya que afecta a nuestra región, como es el caso de la enfermedad de Chagas. Asimismo, y por primera vez, nos atrevimos a proponer recomendaciones para la prevención de la MS en la población pediátrica, con lo complejo y relevante que este tema resulta para la práctica clínica pediátrica.
El objetivo es entregar un documento que el médico (cardiólogo, clínico, pediatra o electrofisiólogo) pueda utilizar como herramienta para encontrar la información indispensable para la toma de decisiones. También el colega podrá encontrar pautas para seleccionar la mejor estrategia en la estratificación del riesgo y las recomendaciones terapéuticas para la prevención adecuada de la MS.
En cada uno de los temas del Consenso se expresa la clase de recomendación de acuerdo con la siguiente clasificación:
- Clase I: condiciones para las cuales existe evidencia y/o acuerdo general en que el procedimiento o tratamiento es beneficioso, útil y efectivo.
- Clase II: condiciones para las cuales existe evidencia conflictiva y/o divergencias de opinión acerca de la utilidad/eficacia del procedimiento o tratamiento.
- Clase IIa: el peso de la evidencia/opinión es a favor de la utilidad/eficacia.
- Clase IIb: la utilidad/eficacia está menos establecida por la evidencia/opinión.
- Clase III: condiciones para las cuales existe evidencia y/o acuerdo general en que el procedimiento o tratamiento no es útil/efectivo y en algunos casos puede llegar a ser perjudicial.
Asimismo, si bien en muchos de los temas no existe bibliografía adecuada, se hace referencia al nivel de evidencia sobre la cual se basa la recomendación consensuada de acuerdo con el siguiente esquema de clasificación:
- Nivel de evidencia A: evidencia sólida, proveniente de estudios clínicos aleatorizados o metaanálisis. Múltiples grupos de poblaciones en riesgo (3-5) evaluados. Consistencia general en la dirección y la magnitud del efecto.
- Nivel de evidencia B: evidencia derivada de un solo estudio clínico aleatorizado o grandes estudios no aleatorizados. Grupos limitados (2,3) de poblaciones en riesgo evaluadas.
- Nivel de evidencia C: consenso u opinión de expertos y/o estudios pequeños, estudios retrospectivos, registros.
Por lo comentado hasta aquí, aspiramos a que este completo Consenso realmente se transforme en un texto de referencia para todos los cardiólogos y en una guía para la toma de decisiones.
CAPíTULO 1
Síndromes de intervalo QT prolongado
y corto
Los síndromes de intervalo QT prolongado hereditarios
Los síndromes de intervalo QT prolongado (QTL) hereditarios son canalopatías que prolongan la repolarización ventricular y predisponen al síncope y/o a la MS (1). Los síncopes se deben a una variedad peculiar de taquicardia ventricular (TV) polimórfica cuya configuración electrocardiográfica le valió la denominación de torsade de pointes (2).
Estas arritmias suelen ser autolimitadas, pero en ocasiones pueden degenerar en fibrilación ventricular (FV) y producir la MS del paciente. Se desencadenan por estrés físico o mental y la natación, en algunas familias pueden ocurrir en reposo o incluso durmiendo (3). En condiciones fisiológicas, la duración del intervalo QTc depende de la edad y el sexo (tabla 1) (4).

Es clásica la descripción de tres síndromes de intervalo QT prolongado hereditarios:
1. El síndrome de Jervell y Lange-Nielsen (JLN)
En 1957, Jervell y Lange-Nielsen describieron a una familia con seis niños de corta edad con sordera congénita; cuatro con síncopes recurrentes, tres de los cuales fallecieron de manera repentina y sus electrocardiogramas mostraban un intervalo QT muy prolongado (5).
2. El síndrome de Romano-Ward
El síndrome de Romano-Ward se observa con mayor frecuencia que el anterior y se diferencia en que la audición es normal y en general la herencia es autosómica dominante (6). A pesar de ello existe una proporción mayor de mujeres afectadas; una de las causas sería que las madres portadoras de las mutaciones genéticas transmiten con mayor frecuencia la enfermedad a sus hijas (7).
En el síndrome de Romano-Ward se describieron 12 mutaciones cromosómicas que involucran a los canales iónicos o a las proteínas de la membrana celular que regulan los intercambios de sodio, potasio y calcio (3). La incidencia se estima en un caso por cada 2.500 a 5.000 individuos de la población general y causa entre 3.000 y 4.000 muertes anuales en niños y adolescentes (6).
3. La variedad esporádica
En 25% a 30% de los casos con fenotipo del síndrome de QTL y audición normal no se pueden identificar mutaciones genéticas conocidas y carecen de un carácter familiar identificable (6).
Recomendaciones diagnósticas
Diagnóstico de los síndromes de QTL hereditarios (7)
Clase I
1. Mapeo genético positivo. (Nivel de evidencia A.)
2. Criterios de Schwartz y colaboradores, puntaje ³ 4. (Nivel de evidencia B.)
El diagnóstico de certeza se realiza por la demostración de la alteración cromosómica responsable en el estudio genético. No obstante, hasta el momento, las mutaciones conocidas se hallan solo en 70% a 85% de los casos con fenotipo positivo (8). La exploración genética negativa en familiares de pacientes con mutaciones cromosómicas conocidas e identificadas descarta el diagnóstico de la afección (9). Cuando no se dispone del estudio genético se utilizan las pautas propuestas por Schwartz y colaboradores (10) que se basan sobre criterios electrocardiográficos, clínicos y familiares, además de la duración del intervalo QTc (tabla 2).

Para obtener el puntaje total, cuando se consideran los episodios de torsade de pointes deben excluirse los síncopes, y de los criterios de la historia familiar se toma en cuenta uno solo. De este modo, el puntaje máximo es 9.
Si el puntaje es ³ 4, la probabilidad diagnóstica de padecer la afección es alta y tiene una sensibilidad de 24% y una especificidad de 99% (3).
Algunos autores utilizan los criterios de Keating (11), que diagnostica el síndrome de QTL hereditario familiar; en los sintomáticos, cuando ese intervalo es ³ 450 ms y en los asintomáticos, cuando el intervalo QTc es ³ 470 ms con una sensibilidad de 36% y una especificidad de 99%.
Otro hecho para destacar es la necesidad de realizar varias mediciones por las variaciones espontáneas que se observan en el intervalo QTc (3).
Familias con síndrome de intervalo QT prolongado y riesgo de muerte súbita
Los pacientes con síndrome QTL pueden presentar eventos cardíacos arrítmicos de manera independiente de la duración del intervalo QTc y de la alteración cromosómica involucrada. Además, existen modificadores genéticos adicionales como el polimorfismo del gen NOS1AP, que tendría un papel determinante en el riesgo arrítmico que presentan los portadores de la enfermedad (12).
El riesgo se considera muy elevado, elevado, intermedio o bajo (7).
Riesgo muy elevado
1. Pacientes recuperados de un paro cardíaco.
2. Síndrome de Jervell y Lange-Nielsen.
3. QTc ³ 0,60 s.
4. Pacientes con síncopes recurrentes a pesar de los betabloqueantes.
Riesgo elevado
1. Pacientes con QTL 2 o 3 y QTc ³ 500 ms.
2. Portadores del polimorfismo en el gen NOS1AP.
3. QTL 2 con mutaciones en la región del poro.
Riesgo intermedio
1. QTL 1 con betabloqueantes, asintomáticos y QTc ³ 500 ms.
Riesgo bajo
1. Pacientes asintomáticos con QTc £ 500 ms y con betabloqueantes.
Recomendaciones terapéuticas
Tratamiento agudo de las torsade de pointes
Los episodios de torsade de pointes que provocan síncope y/o detención cardiocirculatoria deben ser tratados con (13):
Clase I
A. Cardioversión eléctrica inmediata. (Nivel de evidencia A.)
Clase IIa
A. Corregir y/o eliminar cualquier otro factor intercurrente que pueda agravar el cuadro (hipopotasemia, fármacos, etcétera). (Nivel de evidencia B.)
B. Sulfato de magnesio, 2 gramos por vía intravenosa. (Nivel de evidencia B.)
Clase IIb
A. Marcapaseo ventricular transitorio. (Nivel de evidencia C.)
Tratamiento crónico
El manejo de los síndromes de intervalo QT prolongado hereditarios se basa en pilares terapéuticos paliativos, destinados a prevenir las arritmias ventriculares malignas y la MS (3-7).
Clase I
A. Cambios de estilo de vida. (Nivel de evidencia B.)
B. Fármacos betabloqueantes. (Nivel de evidencia A.)
C. Fármacos betabloqueantes + cardiodesfibrilador implantable (CDI) en pacientes recuperados de un paro cardíaco. (Nivel de evidencia A.)
Clase IIa
A. Fármacos betabloqueantes + CDI en pacientes con síncopes recurrentes a pesar de los betabloqueantes. (Nivel de evidencia B.)
Clase IIb
A. Fármacos betabloqueantes + CDI en pacientes de riesgo alto. (Nivel de evidencia B.)
B. Marcapasos DDD. (Nivel de evidencia B.)
C. Resección de la cadena ganglionar parasimpática izquierda. (Nivel de evidencia B.)
Los agentes betabloqueantes son particularmente efectivos en los portadores de mutaciones que involucran a los canales del potasio y de manera primordial a los que tienen como disparador de los eventos arrítmicos el incremento del tono adrenérgico. Los betabloqueantes de elección son el propranolol (3 a 10 mg/kg de peso) y el nadolol (80 a 160 mg/día). En pacientes con afecciones respiratorias se puede utilizar el metropolol. El CDI es aconsejable en los casos con historia familiar de MS, en los recuperados de un paro cardiorrespiratorio por FV y/o TV que no son motivados por causas transitorias y/o reversibles. En los que continúan experimentado síncopes por episodios de TV polifocal o por FV a pesar del tratamiento con dosis adecuada de betabloqueantes (14). Cuando los betabloqueantes no se toleran bien y/u ocasionan bradicardia sinusal sintomática o bien favorecen los episodios de torsade de pointes bradicárdico-dependientes (“variante sódica”), es necesario implantar un marcapasos definitivo. En pacientes con síndrome de intervalo QTL hereditario se aconseja el implante de un marcapasos DDD en los que presentan episodios de TV sostenida pausa-dependientes o en los que las taquiarritmias recurren a pesar de la medicación (15). El marcapasos preferible es el de doble cámara y la frecuencia de estimulación debe ser regulada (en general > 80 lpm) para mantener el intervalo QTc en valores £ 0,44 segundos; no deben superarse los 100 lpm basales para evitar la miocardiopatía por taquicardia.
Seguimiento de los pacientes
con síndrome de intervalo QT prolongado
Los pacientes deben ser evaluados en forma periódica con pruebas de esfuerzo y electrocardiogramas ambulatorios (Holter) de 24 horas. En la mayoría de los casos debe prohibirse la actividad física competitiva.
CAPíTULO 2
Síndrome de Brugada
El síndrome de Brugada (SB) es una enfermedad hereditaria que se caracteriza por la presencia de un patrón electrocardiográfico (ECG) típico de elevación del segmento ST en las derivaciones precordiales derechas asociado con un riesgo incrementado de padecer MS secundaria a arritmia ventricular, en ausencia de cardiopatía estructural demostrable mediante estudios diagnósticos convencionales (16). Los defectos genéticos conocidos se localizan en el cromosoma 3 y afectan el gen que codifica del canal del sodio (SCN5A) (17,18). La susceptibilidad para el desarrollo de arritmias es secundaria a una acentuada dispersión transmural de la repolarización, con la aparición de una ventana vulnerable en la cual un impulso prematuro es capaz de desencadenar arritmias por reentrada.
La prevalencia estimada es inferior a cinco en 10.000 y existe un franco predominio en el sexo masculino (8:1), aunque el carácter dinámico de los hallazgos electrocardiográficos conspira contra el conocimiento de la real prevalencia de la enfermedad. Es la causa más frecuente de MS en menores de 50 años en ausencia de cardiopatía. El ECG típico (tipo 1) muestra elevación del punto J y del segmento ST en precordiales derechas (> 0,2 mV) seguido de ondas T negativas. El diagnóstico definitivo de SB se realiza en presencia de patrón tipo 1 en una derivación precordial de V1 a V3, asociado con alguno de los siguientes hallazgos: FV documentada, TV polimórfica, historia familiar de MS en menores de 45 años, patrón tipo 1 en familiares, inducción de TV con estimulación programada o respiración agónica nocturna. Ante la sospecha o duda, el patrón tipo 1 puede diagnosticarse colocando las derivadas precordiales dos espacios intercostales superiores. El patrón tipo 2, cuya característica es una elevación del segmento ST ³ 2 mm al inicio, una caída con ST ³ 1 mm, seguido de una T positiva o bifásica, y el patrón tipo 3, con apariencia similar a los tipos 1 y 2, aunque con elevación del ST < 1 mm, no son diagnósticos del SB. El hallazgo de alguno de estos últimos patrones tiene importancia fundamentalmente en pacientes sintomáticos y/o con antecedentes familiares, ya que la inducción de un patrón tipo 1 mediante maniobras farmacológicas (ajmalina, flecainida, entre otras) confirma el diagnóstico en pacientes sospechosos (19). En pacientes con patrón tipo 1, la realización de estas pruebas no agrega información y puede ser peligroso. La prueba debe ser realizada por personal experimentado y en un medio seguro.
Estudios de seguimiento han demostrado que los pacientes sintomáticos que presentan un patrón electrocardiográfico típico e historia familiar de MS (HFMS) tienen un riesgo alto de MS. Por el contrario, los pacientes asintomáticos y sin HFMS tienen un riesgo bajo de presentar eventos arrítmicos. La estratificación del riesgo y el abordaje terapéutico en estos pacientes es todavía tema de controversia.
Finalmente, diferentes fármacos han demostrado que son inductores del patrón electrocardiográfico tipo 1 asociado con SB y arritmia ventricular. Por este motivo, los pacientes con SB deberían ser advertidos de no utilizar determinados fármacos debido a su potencial nocivo y, en caso de utilizarlos, que sea en forma controlada (20).
Recomendaciones terapéuticas
Clase I
1. Se debería indicar un CDI en pacientes con SB y antecedentes de un episodio de MS sin otra causa aparente. (Nivel de evidencia C.)
2. Se debería indicar un CDI en pacientes con SB con elevación espontánea del segmento ST-T en V1, V2, o V3 y con antecedentes de síncope de origen cardíaco. (Nivel de evidencia C.)
3. Se debería indicar un CDI en pacientes con SB con TV documentada no causante de MS. (Nivel de evidencia C.)
Clase IIa
1. El implante de un CDI es razonable en pacientes con SB con elevación inducida por fármacos del segmento ST-T y con antecedentes de síncope de origen cardíaco. (Nivel de evidencia C.)
2. El implante de un CDI es razonable en pacientes con SB asintomáticos con elevación espontánea del segmento ST-T y con taquicardia o FV inducida durante un estudio electrofisiológico (EEF), informado el paciente del riesgo del tratamiento y de la probabilidad de beneficio. (Nivel de evidencia C.)
3. El isoproterenol y la quinidina pueden ser útiles para el tratamiento de tormentas eléctricas en pacientes con SB. Esta última puede considerarse para el tratamiento crónico en pacientes sintomáticos en regiones en las que no puede accederse a un CDI. (Nivel de evidencia C.)
Clase IIb
1. El implante de un CDI es razonable en pacientes con SB asintomáticos con elevación inducida del segmento ST-T e historia familiar de MS y con taquicardia o FV inducida durante un EEF. (Nivel de evidencia C.)
Clase III
1. Los fármacos antiarrítmicos del tipo Ic (por ejemplo, flecainida y propafenona) y los de clase Ia con excepción de la quinidina (por ejemplo, procainamida o ajmalina) están contraindicados en pacientes SB. (Nivel de evidencia C.)
Recomendaciones para el manejo clínico de pacientes con síndrome de Brugada
Clase I
1. Se debería informar al paciente portador de patrón de SB sobre la acción de fármacos que puedan favorecer la aparición de arritmias. (Nivel de evidencia C.)
Clase IIa
1. La realización de un EEF con estimulación ventricular programada podría ser de utilidad para estratificar el riesgo en pacientes asintomáticos con patrón de Brugada tipo 1 espontáneo. (Nivel de evidencia C.)
2. Es razonable realizar una monitorización estrecha de una elevación espontánea del segmento ST en pacientes con elevación del ST solamente inducido por fármacos. (Nivel de evidencia C.)
Clase IIb
1. La realización de un EEF con estimulación ventricular programada podría ser de utilidad para estratificar el riesgo en pacientes asintomáticos con patrón de Brugada tipo 1 inducido por fármacos y con historia familiar de MS. (Nivel de evidencia C.)
CAPíTULO 3
Displasia arritmogénica
del ventrículo derecho
La displasia arritmogénica del ventrículo derecho (DAVD) es una miocardiopatía que se caracteriza por una sustitución progresiva, parcial o masiva, del miocardio del VD por tejido adiposo o fibroadiposo.
La prevalencia mundial de esta enfermedad se calcula en 1/2.000 a 1/10.000 según la población estudiada; es más común en algunas regiones de Italia, como la del Veneto. La DAVD es la causa más frecuente de MS en atletas de Italia y podría corresponderle hasta 20% de las MS en adultos jóvenes (21). En otros países, como Estados Unidos, correspondería a 5% de dichas muertes en pacientes menores de 65 años (22). Las anomalías estructurales progresivas, que consisten en el reemplazo fibroadiposo, serían el sustrato arritmogénico de esta enfermedad. Las taquiarritmias responsables se producirían por reentradas originadas en el VD. Estas taquicardias son típicamente muy rápidas, de carácter paroxístico y con recurrencias frecuentes.
Algunos autores han demostrado que los pacientes portadores de la enfermedad tienen un riesgo elevado de MS. En una serie de necropsias de jóvenes fallecidos súbitamente se observó una incidencia elevada de DAVD (23); sin embargo, otros autores han observado un riesgo bajo de MS en el seguimiento a largo plazo (24). La tasa de mortalidad anual sin tratamiento es de 2,5%-3% y en individuos en tratamiento es de 1% (25). La terapéutica en esta patología intenta prevenir fundamentalmente los eventos letales. El CDI mejora la sobrevida de los pacientes en prevención secundaria y en aquellos con riesgo aumentado de MS, por lo que estaría indicado en este tipo de pacientes. No obstante, el tratamiento inicial se realiza habitualmente con betabloqueantes; si estos resultan inadecuados para el control de los síntomas o la prevención de recurrencias de TV, son necesarios otros antiarrítmicos, como el sotalol y la amiodarona (26), especialmente útiles si logran suprimir la inducibilidad de la arritmia (27). Hay poca experiencia con ablación y por los resultados iniciales no parece que sea muy efectiva, aunque nuevos estudios con los métodos de mapeo tridimensional son más optimistas y brindan mejores resultados, por lo que sería una alternativa para considerar en los casos que experimentan recurrencias a pesar del uso de antiarrítmicos, para casos con múltiples choques del CDI o en los pacientes que no pueden recibir un CDI (28).
Prevención secundaria
Recientes trabajos multicéntricos han demostrado consistentemente una frecuencia alta de choques apropiados y una tasa muy baja de muerte arrítmica en pacientes con DAVD tratados con CDI (29), lo que lo transforma en la terapia de elección en estos casos. Si bien no existen trabajos aleatorizados prospectivos que evalúen terapia farmacológica versus CDI en estos pacientes para la prevención secundaria de MS, estudios no aleatorizados y la opinión de expertos apoyan la utilización de los dispositivos por sobre el uso de fármacos.
Prevención primaria
Aunque algunos autores sugieren que los pacientes que reúnen los criterios diagnósticos de DAVD tienen un riesgo alto de MS y ello sería suficiente para indicar un CDI, aún se requieren más estudios para confirmarlo (30).
Cuando se piensa en prevención primaria, se debe tener en mente que no existen marcadores de riesgo clínicos bien definidos en DAVD, ya que los trabajos de sobrevida con CDI y prevención primaria en esta patología no son muy numerosos.
En los pacientes de riesgo alto, la tasa de choques apropiados del CDI es de 10% al año, mientras que en los pacientes con riesgo bajo la tasa de choque es muy inferior. Por ello, los pacientes con diagnóstico de DAVD asociada con riesgo alto de MS deben ser considerados para terapia con CDI. Algunos expertos han propuesto el uso del CDI basado en la presencia de factores de riesgo de MS, como (31-37):
1. Evidencia de daño del VD extenso.
2. Compromiso del ventrículo izquierdo (VI).
3. Aneurismas del VD.
4. Displasia asociada con un locus del genotipo cromosómico 1q42-43.
Otros factores de riesgo de utilidad clínica que se han identificado son:
- Sexo masculino.
- La detección de TV no sostenida en la monitorización no invasiva.
- La dilatación grave del VD.
- La inducción de TV durante el EEF.
Este último factor es tema de controversia, con resultados dispares. Corrado y colaboradores (29) demostraron que el número de las descargas apropiadas del CDI no difería entre los pacientes en los que se inducía TV y en los que no se inducía TV en el EEF, mientras que Witcher y colaboradores (24) evidenciaron que la inducción de TV o FV en el EEF mostraba una tendencia a presentar un número mayor de choques apropiados. En el mismo sentido, para Roguin y colaboradores (33), la inducción de TV fue el principal predictor independiente de choques apropiados del CDI.
La importancia de identificar a los subgrupos de riesgo alto que tienen indicación de CDI radica en que se logre optimizar la relación riesgo-beneficio, dado que, a los efectos no deseables generales con el uso de CDI como infecciones o choques espurios, en esta patología específica se agregan dos cuestiones muy importantes:
1. La infiltración adiposa podría dificultar la obtención de umbrales adecuados y sobre todo la posibilidad de un buen sensado con la consecuencia de una falta de detección de las arritmias potencialmente letales.
2. El VD está adelgazado y no contráctil, por lo que, si bien es una complicación poco frecuente, existe el riesgo de perforación y taponamiento cardíaco durante la colocación del catéter.
Recomendaciones
Clase I
1. El implante del CDI se recomienda para la prevención de la MS en pacientes con DAVD con TV sostenida, FV documentada o síncope de probable origen arrítmico y que tienen una expectativa de vida superior a un año. (Nivel de evidencia B.)
Clase IIa
1. El implante del CDI puede ser efectivo para la prevención de MS en pacientes con DAVD con factores de riesgo para MS y que tienen una expectativa de vida superior a un año. (Nivel de evidencia C.)
2. El tratamiento farmacológico con betabloqueantes y/o antiarrítmicos (sotalol o amiodarona) estaría indicado en los pacientes con DAVD sin factores de riesgo para MS o en pacientes con DAVD y factores de riesgo para MS en los que no es posible el implante del CDI. (Nivel de evidencia C.)
3. La ablación puede ser útil como terapia adyuvante en pacientes con DAVD y TV recurrente a pesar de la terapia antiarrítmica óptima. La indicación estaría dada por la presencia de TV incesante, descargas frecuentes del CDI o la imposibilidad de recibir un CDI o fármacos. (Nivel de evidencia C.)
Clase IIb
1. El EEF podría ser útil para la estratificación del riesgo de MS en pacientes con DAVD. (Nivel de evidencia C.)
CAPíTULO 4
Miocardiopatía hipertrófica
La miocardiopatía hipertrófica (MCH) es una entidad que se caracteriza por la presencia de hipertrofia ventricular izquierda no atribuible a otras causas, como enfermedad valvular o hipertensión arterial (38).
Es la cardiopatía genética más frecuente (1/500 nacimientos), de transmisión autosómica dominante, con diversidad intragénica importante; se han identificado más de 600 mutaciones en diferentes genes que codifican proteínas del sarcómero (39). Por otra parte, existen también otras enfermedades en las que interactúan causas genéticas y metabólicas y tienen una manifestación fenotípica similar (fenocopias), como es el caso de la enfermedad de Anderson-Fabry, de las glucogenosis y de algunas enfermedades mitocondriales (39).
Muchos pacientes son diagnosticados durante exámenes de rutina y en algunos casos la MS puede ser la primera manifestación de la enfermedad. Los síntomas suelen ser la disnea de esfuerzo, angor, presíncope y síncope. En estos pacientes predomina la disfunción diastólica y habitualmente presentan obstrucción al tracto de salida del VI (38). Durante la evolución de la enfermedad, algunos pacientes pueden presentar progresión de los síntomas, fibrilación auricular (FA), fenómenos embólicos y, en un porcentaje muy bajo, insuficiencia cardíaca (IC) por disfunción sistólica. También debemos destacar que esta población tiene un riesgo mayor de desarrollar endocarditis infecciosa, sobre todo en las formas obstructivas y en los que tienen implantados dispositivos intracavitarios como CDI y/o marcapasos (38).
Evaluación del riesgo de muerte súbita
Nuestro gran desafío es poder identificar a los pacientes que tienen más riesgo de padecer un episodio de MS. La incidencia de MS en la MCH es de hasta 6% en centros de atención terciaria y menor de 1% en poblaciones no seleccionadas. La MCH es la principal causa de MS en quienes practican deportes a nivel competitivo y, si bien es más frecuente en los jóvenes, puede ocurrir a cualquier edad (38).
A continuación se resumen los principales factores de riesgo mayores de MS que fueron incluidos en el Consenso Internacional de MCH publicado en 2003 y dentro de los “factores menores” hemos decidido agregar también otros predictores sobre la base de diferentes publicaciones aparecidas desde 2003 hasta la fecha (38-40).
Factores de riesgo mayores
- Paro cardíaco por FV documentada.
- TV sostenida espontánea.
- Historia familiar de MS prematura.
- Síncope reciente de origen inexplicable.
- Hipertrofia ventricular extrema (³ 30 mm).
- TV no sostenida (repetitivos o prolongados con FC ³ 120 lpm).
Factores de riesgo menores (posibles en determinados pacientes)
- Fibrilación auricular.
- Isquemia miocárdica.
- Obstrucción al tracto de salida del VI (> 30 mm Hg).
- Las mutaciones genéticas de riesgo alto.
- Pacientes jóvenes.
- Respuesta anormal de la presión arterial durante el ejercicio.
- Disfunción sistólica con fracción de eyección < 50%.
- Enfermedad coronaria asociada.
- Puentes musculares.
- Pacientes a los que se les practicó ablación septal con alcohol.
- Evidencia de fibrosis miocárdica.
Debemos aclarar que el EEF para inducción de taquiarritmias ventriculares no es un criterio y no debe utilizarse como marcador de riesgo.
Alternativas terapéuticas para los pacientes con miocardiopatía hipertrófica y riesgo alto de muerte súbita
Se han evaluado numerosos fármacos para prevenir la MS en pacientes con MCH, pero ninguno ha demostrado que sea efectivo para disminuir el riesgo (38). Si bien se ha comunicado que 25% de los pacientes que presentaron choques apropiados por un CDI recibían amiodarona, dicho tratamiento está contemplado cuando el CDI no está disponible o es rechazado por el paciente (41). El CDI es el único tratamiento que ha demostrado gran eficacia para prevenir la MS en los pacientes con más riesgo.
Indicaciones de CDI en la miocardiopatía hipertrófica
Prevención secundaria
En pacientes resucitados de MS y en los sintomáticos por TV y/o síncope vinculado con arritmia ventricular (38-41).
Prevención primaria
La indicación de un CDI como prevención primaria difiere considerablemente según el país de origen, el sistema de salud, la accesibilidad a los dispositivos y las opiniones de diferentes expertos (40). Unos de los puntos de mayor controversia en la actualidad es si es suficiente un solo factor de riesgo para indicar un CDI o si los pacientes deben reunir dos o más criterios (40,41). La primera postura es la que predomina entre los autores de Estados Unidos, quienes sostienen que un solo factor de riesgo puede ser suficiente para indicar el implante de un CDI como prevención primaria en determinados pacientes y consideran que no existe relación entre el número de predictores de riesgo y la MS. Demostraron también que el tiempo desde el implante del CDI hasta el primer choque apropiado puede llegar a ser de hasta diez años y también que un tercio de los pacientes en prevención primaria que recibieron choques apropiados tenían un solo factor de riesgo (40). Sin embargo, es importante destacar que en algunos de los estudios en los que se apoya dicha estrategia no se les efectuó Holter de rutina a los pacientes.
Por su parte, autores europeos afirman que el pronóstico es más ominoso cuando se asocian dos o más factores de riesgo. De acuerdo con la evidencia y las recomendaciones de diferentes expertos, nosotros hemos rescatado los elementos más importantes de las dos principales posturas a nivel internacional y desarrollamos una guía para seleccionar a los pacientes que más se pueden beneficiar con esta terapéutica (tabla 3).
CAPíTULO 5
Cardiopatía isquémica
Prevención primaria
Diversos estudios han evaluado la utilidad del uso del CDI en prevención primaria de mortalidad en pacientes con cardiopatía isquémica. En la tabla 4 se resumen los aspectos más relevantes de esos estudios.
El estudio MADIT I (42) fue el primero en demostrar la utilidad de los dispositivos en prevención primaria. Se incluían pacientes postinfarto con FEVI < 36% y TV no sostenida, sin indicación de revascularización. Estos pacientes eran llevados a EEF y si se les inducían arritmias ventriculares sostenidas que no se suprimían con procainamida se les implantaba un CDI o seguían tratamiento convencional. Se reclutaron 196 pacientes (95 CDI y 101 tratamiento convencional) y durante un seguimiento promedio de 27 meses, la terapia con CDI se asoció con una disminución significativa de la mortalidad de 54% (15 muertes en el grupo CDI y 39 en el grupo tratamiento convencional; p = 0,009). El estudio MADIT recibió numerosas críticas (pequeño número de pacientes, tiempo de seguimiento prolongado y diferencias entre grupos en el tratamiento instaurado), pero fue un trabajo pionero en prevención primaria.
El estudio CABG-PATCH (43) evaluó el efecto del implante de un CDI epicárdico en el momento de la revascularización miocárdica quirúrgica en pacientes con FEVI £ 35% y anomalías en el ECG de señal promediada. Luego de cuatro años de seguimiento no se observó beneficio con la terapia con CDI.
El estudio MUSTT (44) incluyó a pacientes parecidos a los del MADIT; en un grupo se realizaba tratamiento antiarrítmico guiado por EEF y el otro grupo no recibió tratamiento. El seguimiento fue de 60 meses. El tratamiento guiado por el EEF redujo significativamente la mortalidad arrítmica y cardíaca a los 24 y a los 60 meses de seguimiento. Ese beneficio se atribuyó al uso del CDI, que fue implantado en 58% de estos últimos pacientes. El registro MUSTT (45) mostró una mortalidad alta a los cinco años en los pacientes sin arritmia ventricular inducible, la cual fue ligeramente inferior a la de aquellos inducibles, pero significativamente más alta que la del grupo tratado con CDI. Esto sugiere que estos pacientes podrían haberse beneficiado con un CDI y que la estimulación ventricular programada es un mal estratificador de riesgo en este tipo de pacientes.
El estudio MADIT II (46) fue un trabajo prospectivo, que incluyó pacientes que habían sufrido un infarto agudo de miocardio (IAM) más de un mes atrás con una FEVI £ 30%. Una rama se asignó a CDI y la otra a tratamiento convencional, en una proporción 3:2. El objetivo primario del estudio fue muerte por cualquier causa. Se incluyeron 1.232 pacientes que fueron seguidos en promedio durante 20 meses. El estudio se suspendió prematuramente por alcanzar el objetivo de eficacia del CDI, con una tasa de mortalidad total de 14,2% en el grupo CDI versus 19,8% en el grupo con tratamiento convencional (HR 0,69, IC 95% 0,51-0,93; p = 0,016).
El estudio DINAMIT (47) evaluó el implante de un CDI precozmente luego de un infarto (6-40 días, media 18 días). Si bien se obtuvo una reducción significativa de la muerte arrítmica con el CDI (HR 0,42; p = 0,009), la mortalidad total fue similar en ambos grupos de tratamiento (p = 0,66). Esto se debió a un aumento significativo de la muerte no arrítmica (HR 1,75; p = 0,02).
El estudio SCD-HeFT (48) enroló a 2.521 pacientes con disfunción ventricular izquierda (FEVI £ 35%) de etiología isquémica y no isquémica que estuvieran en IC CF II-III, a pesar de recibir tratamiento médico óptimo. Los pacientes fueron aleatorizados a tres grupos de tratamiento: CDI unicameral (n = 829), amiodarona (n = 845) y placebo (n = 847). Luego de un período de seguimiento de 45 meses, 666 pacientes (26,4%) alcanzaron el punto final primario (mortalidad total): 244 pacientes en el grupo placebo (29%), 240 en el grupo amiodarona (28%) y 182 en el grupo CDI (22%). Comparada con placebo, la terapia con CDI se asoció con una reducción relativa significativa de la mortalidad de 23% (HR 0,77; IC 95% 0,62-0,96; p = 0,007), mientras que la terapia con amiodarona no brindó beneficio clínico. La disminución del riesgo de muerte obtenida con el CDI fue comparable en el subgrupo con etiología isquémica y no isquémica y estuvo concentrada en pacientes con IC CF II y en aquellos con FEVI £ 30%.
Conclusiones
Los estudios de prevención primaria en cardiopatía isquémica MADIT II y SCD-HeFT han mostrado una disminución de la mortalidad total en dicha población. La simplicidad de los criterios de inclusión, básicamente la fracción de eyección y el IAM previo, han llevado a amplificar de manera notable la indicación de un tratamiento de alto costo como es el implante de un CDI. No obstante, varios autores han cuestionado sus resultados y enfatizan en la necesidad de identificar subgrupos en los que el beneficio sea mayor, así como reconocer a aquellos pacientes con mayor mortalidad no arrítmica en quienes el dispositivo no ofrece mejoría en la sobrevida y, de esta manera, optimizar la ecuación costo-beneficio (49,50). Los autores del MADIT II publicaron un puntaje para cuya elaboración seleccionaron cinco marcadores de riesgo: edad > 70 años, QRS > 120 ms, FA o CF > II y BUN entre 26 y 50 mg/dl. El beneficio del CDI solo se apreció en los que reunían una o dos variables (tabla 5) (51).
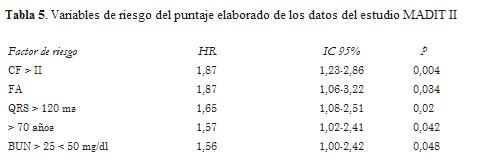
Los datos provenientes de los estudios MADIT I y MUSTT, a pesar de diferencias metodológicas de importancia, son coincidentes en el beneficio de la terapia del CDI en pacientes con criterios de riesgo alto, como la presencia de disfunción grave del VI, TVNS y la inducción positiva de TV.
En lo que respecta a la relación costo-efectividad de la indicación de un CDI en prevención primaria de MS, disponemos de un modelo publicado basado en los resultados del MADIT II (52). Este estudio muestra una relación costo-efectividad inapropiada, con una estimación media del cociente de costo-efectividad incremental de alrededor de 235.000 dólares por año de vida salvado, la cual está claramente fuera del rango aceptable. Este resultado desfavorable se debió en particular al hecho de que la estimación de aumento de la expectativa de vida en estos pacientes con la colocación de un CDI fue de solo dos meses en el período de duración del estudio (3,5 años); según este modelo, solo se obtendría un valor de costo-efectividad razonable para los parámetros de Estados Unidos en el caso de que la reducción en mortalidad con el CDI se mantenga durante 12 años. Aunque no se han publicado datos de este tipo en nuestro país, es improbable que (en el grupo general de pacientes considerado) la relación costo-efectividad sea apropiada en nuestro medio, dado que en general cuanto menor es el PBI per cápita de un país, menos dinero está disponible para utilizar en salud y más restrictivos son los puntos de corte de costo-efectividad. Así, un tratamiento que dudosamente sea costo-efectivo en Estados Unidos, con toda probabilidad será excesivamente caro para ser utilizado en Argentina-Uruguay. Sin embargo, es dable estimar que la relación costo-efectividad podría ser apropiada en nuestro país en aquellos casos en los que la eficacia estimada del CDI fuera mayor, dado que se ha demostrado que la eficacia del CDI es la variable de mayor peso en la determinación de su costo-efectividad (53). De esta manera, la selección de pacientes en los cuales es esperable un beneficio mayor del CDI podría generar una indicación apropiada de CDI en Argentina en términos de costo-efectividad.
Por lo mencionado, en la población post IAM de nuestro medio surge la necesidad de seleccionar grupos de mayor riesgo en los cuales utilizar este valioso recurso terapéutico.
Así, la opinión de este Consenso es que tomar únicamente los parámetros de fracción de eyección y tiempo no es suficiente para indicar un CDI a toda la población post IAM. Por ello, la recomendación de indicación de un CDI en pacientes “símil MADIT II” será de clase IIa.
No obstante, destacamos que dentro de esta población la indicación es más clara en los subgrupos con puntaje 1-2 del MADIT II, en los cuales la relación costo-efectividad será más apropiada. En este aspecto, la duración del complejo QRS > 120 ms resulta una variable estable, fácil de obtener y la que aparece vinculada con mayor beneficio en los pacientes con CDI en los estudios MADIT II y SCD HeFT.
También enfatizamos que subgrupos con comorbilidades serias no son buenos candidatos, en especial aquellos con disfunción renal moderada a grave y con un puntaje de 3 o más del MADIT II debido a la elevada mortalidad no arrítmica en estos pacientes.
Recomendaciones en prevención primaria
Clase IIa
1. Pacientes con > 40 días post IAM, Fey < 30%, CF I o asintomáticos, bajo tratamiento médico óptimo, con expectativa de vida mayor de un año. (Nivel de evidencia A.)
2. Pacientes con > 40 días post IAM, Fey < 35%, CF II-III, bajo tratamiento médico óptimo, con expectativa de vida mayor de un año. (Nivel de evidencia A.)
Clase IIb
1. Pacientes con síncope inexplicado, con cardiopatía estructural, Fey ³ 35% e inducción negativa de TV. (Nivel de evidencia C.)
Clase III
1. Pacientes con < 40 días de evolución post IAM. (Nivel de evidencia A.)
2. Pacientes en CF IV que no tengan indicación concomitante de resincronización o que se encuentren en lista de trasplante. (Nivel de evidencia C.)
3. Pacientes en quienes se planea una revascularización o han sido revascularizados en los últimos tres meses. (Nivel de evidencia C.)
4. Pacientes con comorbilidades serias como EPOC grave, insuficiencia renal grave, alteraciones psiquiátricas graves o expectativa de vida con calidad de vida aceptable menor de un año. (Nivel de evidencia C.)
Prevención secundaria
La población de pacientes que ha sufrido una MS cardíaca reanimada de causa no corregible se constituye en un grupo llamado de riesgo alto por la elevada probabilidad de recurrencia de un nuevo episodio. La prevención secundaria de MS se refiere al tratamiento instaurado para evitar o tratar la repetición de nuevos eventos arrítmicos mayores, como son la FV y la TV sostenida, arritmias amenazantes para la vida.
Un trabajo prospectivo y aleatorizado en prevención secundaria que incluyó a 1.016 pacientes comparó el tratamiento con CDI versus fármacos antiarrítmicos de clase III (predominantemente amiodarona empírica) y demostró mejoría en la sobrevida en los pacientes con CDI (54). Este estudio comunicó una reducción significativa del riesgo relativo (RRR) estimada de 39% (IC 95% 19%-59%) en el grupo CDI al año, de 27% (IC 95% 6%-48%) a los dos años y de 31% (IC 95% 10%-52%) a los tres años. Otros dos estudios prospectivos aleatorizados en un grupo de pacientes con antecedentes de MS mostraron resultados similares (tabla 6) (55,56). En un metaanálisis de los estudios antes mencionados, el CDI se asoció con una RRR significativa, de 50% en la muerte arrítmica y de 25% en la mortalidad por todas las causas (57).
La mayoría de los pacientes incluidos en los trabajos prospectivos aleatorizados de pacientes resucitados de MS (AVID, CIDS, CASH) tenían enfermedad coronaria.
La FEVI promedio tenía un rango de entre el 32% y el 45%. La evidencia actual es robusta y soporta fuertemente el beneficio en la sobrevida de la utilización del CDI versus el tratamiento con fármacos antiarrítmicos en este grupo de pacientes.
Recomendaciones en prevención secundaria
Clase I
1. La terapia con CDI está indicada en pacientes que sobrevivieron a un paro cardíaco debido a FV o TV sostenida con compromiso hemodinámico no atribuibles a causas transitorias o reversibles. (Nivel de evidencia A.)
2. La terapia con CDI está indicada en pacientes con enfermedad coronaria, FEVI menor o igual a 40% y TV sostenida espontánea con o sin compromiso hemodinámico. (Nivel de evidencia B.)
3. La terapia con CDI está indicada en pacientes con síncope de origen desconocido, enfermedad coronaria con FEVI menor o igual a 40%, con TV sostenida con compromiso hemodinámico o FV inducidas en el EEF. (Nivel de evidencia B.)
Clase IIa
1. El implante de un CDI es razonable en pacientes con enfermedad coronaria, TV sostenida con función ventricular normal o levemente deteriorada. (Nivel de evidencia C.)
Clase III
1. El CDI no está indicado en pacientes que no tienen una expectativa de vida razonable aun cumpliendo con los criterios de implante antes mencionados. (Nivel de evidencia C.)
2. El CDI no está indicado en pacientes con taquicardia o fibrilación ventricular incesantes. (Nivel de evidencia C.)
3. El CDI no está indicado en pacientes con enfermedades psiquiátricas importantes pasibles de agravarse con la implantación del dispositivo o que pueden imposibilitar un seguimiento sistemático. (Nivel de evidencia C.)
4. El CDI no está indicado en pacientes en CF IV de la NYHA refractarios al tratamiento farmacológico y que no son candidatos para la terapia de resincronización cardíaca ni para el trasplante cardíaco. (Nivel de evidencia C.)
5. El CDI no está indicado en pacientes con TV tratable mediante ablación por radiofrecuencia o cirugía (por ejemplo, TV del tracto de salida del ventrículo derecho o izquierdo, TV idiopática o fascicular sin cardiopatía estructural. (Nivel de evidencia C.)
CAPíTULO 6
Miocardiopatía dilatada idiopática
La miocardiopatía dilatada (MCPD) idiopática constituye la segunda causa de muerte después de la cardiopatía isquémica en los pacientes con insuficiencia cardíaca. La mortalidad de estos pacientes a los cinco años se estima en alrededor de 20% y aproximadamente 30% corresponden a MS.
El tratamiento con IECA y betabloqueantes ha disminuido la mortalidad en este grupo de pacientes con disfunción ventricular izquierda significativa, además de reducir el número de internaciones. La asociación con otros fármacos, como diuréticos del asa o antagonistas de la aldosterona, ha probado que disminuye aun más la mortalidad en este grupo de pacientes cuando se encuentran sintomáticos con IC en CF III-IV. A pesar del tratamiento médico óptimo en estos pacientes sigue existiendo un riesgo aumentado de MS. En la mayoría de los estudios aleatorizados a gran escala, más de la mitad de los pacientes incluidos son portadores de MCPD secundaria a enfermedad coronaria y en ellos el CDI se asoció con una mejora de la sobrevida en comparación con el uso de fármacos antiarrítmicos. Esto se ha demostrado tanto en prevención primaria como secundaria.
Son pocos los estudios realizados en MCPD idiopática. Entre ellos, el estudio AMIOVIRT y el registro CAT no mostraron beneficios del CDI en este grupo de pacientes en relación con el uso de amiodarona o control (58,59). Ambos incluyeron un número pequeño de pacientes y una población con una mortalidad menor que la esperada, por lo que finalizaron precozmente. El estudio DEFINITE incluyó 458 pacientes con MCPD idiopática, Fey < 35% y arritmia ventricular (60). El implante de un CDI redujo en forma significativa el riesgo de MS. En el estudio SCD-HeFT, los pacientes fueron asignados a tres ramas: placebo, amiodarona o CDI (48). En este estudio, 48% de los pacientes eran portadores de MCPD idiopática y en el grupo CDI se encontró 23% de reducción en la mortalidad total (p = 0,07).
Ante la selección del paciente individual para el implante de un CDI debemos tener en cuenta los factores clínicos y las comorbilidades asociadas. La edad mayor de 80 años, el antecedente de FA, una CF III-IV y valores de creatinina por encima de 1,8 mg/dl son algunas variables asociadas con un aumento de la mortalidad al año postimplante de un CDI (51). La mortalidad aumenta de 0,4% a 21% en los pacientes con menos o más de dos de estas variables, respectivamente.
Recomendaciones
Clase I
1. En los pacientes con MCPD idiopática que han presentado TV sostenida o FV, con síncope o MS debe efectuarse el implante de un CDI. (Nivel de evidencia A.)
2. En pacientes con TV rama a rama y en aquellos en los que se presuma una TV idiopática deben considerarse la realización de un EEF y la eventual ablación. (Nivel de evidencia C.)
Clase IIa
1. En los pacientes con MCPD idiopática, Fey £ 35%, CF II-III de la NYHA estable en los últimos tres meses, con tratamiento óptimo y una expectativa de vida mayor de un año debería implantarse un CDI. (Nivel de evidencia A.)
2. En pacientes con MCPD idiopática, Fey > 35% y TV sostenida refractaria a fármacos antiarrítmicos debe considerarse el implante de un CDI. (Nivel de evidencia C.)
3. En pacientes con síncope inexplicado, MCPD idiopática y deterioro significativo de la Fey debe considerarse el implante de un CDI. (Nivel de evidencia C.)
4. En los pacientes con MCPD idiopática que han presentado un episodio de taquicardia con QRS ancho sería útil la realización de un EEF con fines diagnósticos. (Nivel de evidencia C.)
5. Los pacientes con MCPD idiopática, Fey > 35% y arritmia ventricular no sostenida, asintomática, serían pasibles de tratamiento con antiarrítmicos del grupo II o III. (Nivel de evidencia C.)
6. En los pacientes que han recibido múltiples choques del CDI por TV a pesar de tratamiento médico completo debe considerarse la realización de una ablación con catéteres. (Nivel de evidencia C.)
Clase IIb
1. En los pacientes con MCPD idiopática, Fey £ 35%, CF I de la NYHA, con tratamiento óptimo y una expectativa de vida mayor de un año puede considerarse el implante de un CDI. (Nivel de evidencia C.)
2. En los pacientes con MCPD idiopática, Fey > 35% y TV sostenida puede considerarse el uso de amiodarona. (Nivel de evidencia C.)
CAPíTULO 7
Enfermedad de Chagas
La miocardiopatía de origen chagásico es una de las principales causas de morbilidad en América Latina y particularmente en nuestro país. Se estima que se encuentra presente en 25% a 30% de los pacientes con serología positiva (61). Típicamente produce defectos de la conducción y el automatismo, anormalidades de la motilidad parietal ventricular, insuficiencia cardíaca, fenómenos tromboembólicos y lo que principalmente nos preocupa en este tema: arritmias ventriculares y MS. Se estima que la MS es responsable de alrededor de 55%-65% de las muertes en esta patología y no siempre se encuentra asociada con marcadores clásicos de mortalidad, como el deterioro de la función ventricular (62).
Epidemiología
El porcentaje de individuos infectados que desarrollan lesiones cardíacas en forma crónica varía según las zonas, la edad, el tiempo de exposición en un área endémica, el número de reinfestaciones, el nivel socioeconómico, el tiempo de evolución de la infección y el estado de nutrición (63). El rango de mortalidad depende del estadio de la enfermedad. En la serie de Viotti y colaboradores (64) se observó que en pacientes sin IC, 50% de las muertes totales ocurrieron en forma súbita. En este grupo se observó una mortalidad total de 0,5% en pacientes sin alteraciones del ECG, de 2,8% en aquellos con alteraciones solo en el ECG y de 14% en los que presentaban signos de disfunción ventricular asintomática (diámetros aumentados en el ecocardiograma y/o cardiomegalia en la radiografía de tórax). En pacientes en fase dilatada con IC congestiva se encontró una mortalidad de 50% en el primer año y de 44% de MS por TV y/o FV como causas más frecuentes (61). Un dato relevante y de observación frecuente lo constituyen los pacientes que presentan arritmias ventriculares sostenidas o episodios de MS reanimada, los cuales no tienen evidencias de disfunción ventricular y requirieron un CDI para prevención secundaria. En series recientes que analizaron características y predictores de mortalidad total en pacientes con dichas características, 15%-28% no presentaban disfunción ventricular (65,66).
Estratificación del riesgo
Los principales problemas que debemos enfrentar en la toma de decisiones podrían resumirse en dos cuestiones principales: la inexistencia de ensayos clínicos que incluyan pacientes chagásicos que validen el implante de un CDI y la dificultad de detectar un subgrupo de riesgo alto entre los pacientes que no presentan IC sintomática.
Con respecto al primer problema, diferentes publicaciones de estudios observacionales han demostrado que el deterioro de la función ventricular izquierda, la CF III-IV, la cardiomegalia y la TV no sostenida son indicadores de mal pronóstico en la enfermedad de Chagas crónica (67-69). Estos hallazgos sugerirían que el comportamiento de la cardiopatía chagásica en presencia de disfunción ventricular no sería diferente de las miocardiopatías dilatadas de etiología isquémico-necrótica o idiopática.
En algunos trabajos llama la atención el elevado porcentaje de descargas apropiadas en el seguimiento de poblaciones chagásicas con CDI, con 42% al año y un período corto entre el implante y el primer choque (65,70).
Un desafío mayor lo representan los pacientes sin evidencia clínica de disfunción ventricular, con presencia de ECG anormales por trastornos intraventriculares de la conducción con o sin arritmia ventricular compleja, o aquellos que aún no han sufrido eventos que justifiquen la prevención secundaria. Para ello se ha tratado de determinar distintas maneras de estratificar el riesgo a través de la evaluación de diferentes variables como predictores clínicos combinados o independientes. En esa dirección, Rassi y colaboradores (70) elaboraron un puntaje de riesgo sobre seis factores pronósticos independientes, asignándoles una cantidad de puntos proporcionales a su coeficiente de regresión: CF III-IV 5 puntos; evidencia de cardiomegalia en la radiografía de tórax 5 puntos; disfunción sistólica en el ecocardiograma 3 puntos; TVNS en Holter 3 puntos; QRS de bajo voltaje (< 0,5 mV) 2 puntos, y sexo masculino 2 puntos. Se definieron tres grupos de riesgo: riesgo bajo 0-6 puntos, riesgo intermedio 7-11 puntos y riesgo alto 12-20 puntos. La mortalidad para los grupos fue de 10%, 44% y 84%, respectivamente.
Prevención primaria
Es conocido que no es posible homologar directamente las indicaciones de las guías internacionales a esta patología, ya que no se reclutaron pacientes chagásicos en los ensayos aleatorizados de referencia, por lo cual difícilmente sea aceptada la terapia de CDI de prevención primaria definida solo por variables de función ventricular. Nuestra recomendación en los pacientes con disfunción ventricular asociada con síncope o TV no sostenida es la realización de un EEF y en caso de que tengan TV/FV inducible (independientemente de la tolerancia) se recomienda el implante de un CDI.
En los casos de síncope o de TV no sostenida sin evidencia de disfunción ventricular recomendamos la realización de un EEF; en caso de que induzca TV/FV, independientemente de la tolerancia, se recomienda el implante de un CDI.
Indicaciones de estudio electrofisiológico
Clase I
1. Se indica la realización de un EEF para inducción de TV/FV en pacientes con disfunción ventricular o con discinesias parietales asociadas con síncope no aclarado. (Nivel de evidencia B.)
Clase IIa
1. Se indica la realización de un EEF en los pacientes con síncope no aclarado y/o TVNS sin evidencia de disfunción ventricular. (Nivel de evidencia C.)
2. Se indica la realización de un EEF para inducción de TV/FV en pacientes con disfunción ventricular asociada con TVNS. (Nivel de evidencia B.)
Indicaciones de implante de un CDI
Prevención primaria
Clase I
1. Se indica el implante de un CDI en pacientes con disfunción ventricular o discinesias parietales asociadas con síncope no aclarado con TV/FV inducible en el EEF, con independencia de la tolerancia hemodinámica. (Nivel de evidencia B.)
Clase IIa
1. El implante de un CDI es razonable en pacientes con síncope de causa no aclarada y con disfunción o discinesia ventricular significativa. (Nivel de evidencia C.)
Prevención secundaria
En los casos de prevención secundaria preferimos homologar las indicaciones de las guías internacionales; por lo tanto, aceptamos:
Clase I
1. Se indica el implante de un CDI en los pacientes sobrevivientes de un paro cardíaco secundario a FV o TV con mala tolerancia hemodinámica luego de identificar la causa del episodio y de descartar causas reversibles. (Nivel de evidencia A.)
2. Se indica el implante de un CDI en los pacientes que presentaron TV espontánea con disfunción ventricular (o discinesias parietales asociadas) independientemente de la tolerancia hemodinámica. (Nivel de evidencia B.)
Clase IIb
1. El implante de un CDI es razonable en pacientes con TV sostenida y función ventricular normal. (Nivel de evidencia C.)
CAPíTULO 8
Muerte súbita en pediatría
La incidencia de MS cardíaca debida a enfermedades cardiovasculares en la edad pediátrica es significativamente menor que en la población adulta. La tasa de eventos en niños y en adolescentes es de entre 1,3 y 8,5 muertes cada 100.000 pacientes por año, mientras que en adultos mayores de 35 años es de 100 muertes por cada 100.000 pacientes por año (72). Dada la baja incidencia de eventos, no existen trabajos clínicos aleatorizados para definir la estratificación del riesgo de MS en la población pediátrica, como tampoco se ha definido el papel de las terapias para la prevención primaria. Por lo tanto, el nivel de evidencia para la mayoría de las recomendaciones en pacientes pediátricos es nivel C.
A pesar de estas limitaciones, se han identificado muchos grupos de pacientes jóvenes con un riesgo aumentado de MS en comparación con la población general, incluidos los portadores de enfermedades eléctricas primarias como el síndrome de QT largo congénito (SQTLC), las miocardiopatías y las cardiopatías congénitas (73).
En jóvenes, las causas “reversibles” de MS incluyen el síndrome de Wolff-Parkinson-White, las miocarditis agudas y algunos casos de SQTL inducido por drogas. En muchos pacientes con TV monomórfica en corazón “aparentemente sano”, exámenes más exhaustivos como la angio-RMN cardíaca con contraste o la biopsia endomiocárdica permiten detectar evidencias subclínicas de cardiopatía estructural, de las que la DAVD y las miocarditis son los hallazgos más frecuentes y que además suelen pasar inadvertidas con los métodos no invasivos habituales (74).
Los casos de TV que se desencadenan con el ejercicio tienen mal pronóstico y esta asociación es un marcador sensible de la existencia de un corazón anormal (75). Algunos pacientes con TV polimórfica catecolaminérgica (TVPC) pueden no tener síntomas y a pesar de ello se encuentran en riesgo de MS (76). La arritmia ventricular sintomática puede ser también la manifestación inicial de algunas miocardiopatías.
Hay debate aún respecto del síndrome de MS del lactante (SMSL) y el papel potencial de las arritmias cardíacas como causantes de algunas de estas muertes. Las causas de SMSL están en permanente investigación. Si bien las apneas asociadas con una regulación respiratoria inadecuada es la causa principal, también existen evidencias que señalan que la etiología cardíaca podría explicar 5% del SMSL (disfunción autonómica y arritmias de causa genética) (77).
Las cardiopatías congénitas (CC) representan un espectro diverso de defectos anatómicos y funcionales y por lo tanto existen diferencias significativas respecto de la evolución natural, de la fisiología prequirúrgica y postquirúrgica, así como del riesgo de arritmias y de MS para cada una de ellas. Durante la infancia y la adolescencia, más de 75% de las muertes en pacientes con CC son eventos intrahospitalarios que ocurren en el período perioperatorio. Después de los 20 años, hay un aumento progresivo en la incidencia de MS y de la mortalidad cardíaca total para aquellos con CC operadas.
Las CC con mayor riesgo de MS tardía son la tetralogía de Fallot, la D y L transposición de los grandes vasos (D-TGV y L-TGV), la estenosis aórtica, la anomalía de Ebstein y los distintos tipos de ventrículo único anatómico o funcional. El subgrupo más numeroso es el de pacientes con tetralogía de Fallot, donde el riesgo de padecer MS parece ser dependiente del tiempo. A mayor tiempo de seguimiento, mayor riesgo de MS, que se incrementa si además existen lesiones residuales hemodinámicamente significativas y/o disfunción ventricular. La resolución quirúrgica (o hemodinámica) de estos defectos residuales es prioritaria y debe considerarse antes de iniciar cualquier tipo de tratamiento antiarrítmico.
En general, los pacientes con CC que presentan síncope de causa desconocida o paro cardíaco recuperado deben ser cuidadosamente estratificados. Una respuesta positiva en el EEF puede identificar a pacientes con riesgo alto de MS tardía (78). También se debe considerar la posible existencia de bloqueo auriculoventricular paroxístico, en especial en aquellas patologías que evolucionan espontáneamente a él, como la L-TGV y el canal AV y en los pacientes con el antecedente de bloqueo AV temporario en el período postoperatorio inmediato.
Las anomalías coronarias merecen una mención aparte, ya que son causa de síncope de esfuerzo y MS en los niños mayores y en jóvenes sin antecedentes cardiovasculares conocidos y ECG habitualmente normal. La más común es la anomalía del origen de la coronaria izquierda naciendo del seno de Valsalva derecho. La angulación del ostium coronario o bien la compresión durante el ejercicio intenso de la coronaria izquierda al atravesar la región entre la aorta y la arteria pulmonar pueden causar isquemia miocárdica y el desarrollo de TV o FV. El diagnóstico definitivo se realiza por TAC multicorte o por coronariografía selectiva y tiene indicación de revascularización quirúrgica. La afectación coronaria puede presentarse en forma aislada (fístulas, anomalías de implantación, origen o trayecto) o asociada con CC, como la atresia pulmonar con septum intacto o la hipoplasia del VI. Más raramente se observan aneurismas, dilataciones o estenosis secundarias a enfermedades inflamatorias (Kawasaki, Takayasu, panarteritis nudosa) o arteriosclerosis precoz en las dislipidemias familiares, la hipertensión arterial maligna y el trasplante cardíaco. En estas patologías, si bien los antiarrítmicos con acción vasodilatadora coronaria pueden ser útiles, el tratamiento debe estar dirigido a solucionar la afección coronaria en forma definitiva mediante angioplastia o cirugía de revascularización.
El riesgo de MS en jóvenes con disfunción ventricular grave se ha considerado menor que el de los adultos con similar grado de afectación del VI. Sin embargo, en un estudio de CDI en pacientes jóvenes en espera de trasplante cardíaco, 46% presentó terapias apropiadas en siete meses de seguimiento promedio (79). Hasta tanto no existan datos concluyentes para este grupo etario, el implante de un CDI en prevención primaria debe ser extrapolado de los estudios clínicos aleatorizados de la población adulta para pacientes similares. La decisión de implantar un CDI en prevención primaria en la población pediátrica con riesgo aumentado por el antecedente familiar de MS se plantea frecuentemente en patologías como la MCPH, el SQTLC, la TVPC y el síndrome de Brugada. Los factores de riesgo, si bien son conocidos, aún no han sido validados en estudios clínicos de seguimiento. El riesgo de MS en niños con SQTLC y MCPH es de 2% y 3% por año, respectivamente (80,81). Por otro lado, en pacientes pediátricos sobrevivientes de MS, se ha comunicado que el riesgo a uno y tres años para eventos recurrentes (choque apropiado del CDI) es de 30% y 55%, respectivamente (82).
Recomendaciones
Clase I
1. La terapia con CDI está indicada en todo paciente pediátrico que sobrevivió a un paro cardíaco, en quien se descartó causa corregible, siempre que el paciente reciba tratamiento médico óptimo y tenga una expectativa de vida mayor de un año. (Nivel de evidencia B y C.)
2. La terapia con CDI está indicada en los pacientes pediátricos que presentaron TV sostenida con repercusión hemodinámica asociada con una CC cuando no se les pueda ofrecer un tratamiento alternativo (ablación o cirugía cardíaca) y tengan una expectativa de vida mayor de un año. (Nivel de evidencia B y C.)
3. La terapia con CDI está indicada en pacientes con riesgo alto de MS o arritmias ventriculares graves en asociación con una afectación genética (canalopatías o miocardiopatías) que no pueden ser protegidos adecuadamente por otros métodos (fármacos, ablación por catéter, cirugía cardíaca, marcapasos, estelectomía izquierda, etcétera). (Nivel de evidencia B y C.)
4. Se debe realizar una evaluación hemodinámica mediante cateterismo cardíaco y/o RMN y/o EEF en el paciente pediátrico con TV sostenida para descartar afectación cardíaca subclínica. (Nivel de evidencia C.)
5. La pesquisa familiar de los pacientes pediátricos con sospecha de alteraciones genéticas debe comprender evaluación clínica, ECG, ecocardiograma, Holter, ergometría si la edad lo permite y eventualmente pruebas farmacológicas. (Nivel de evidencia C.)
Clase IIa
1. La terapia con CDI es razonable para pacientes pediátricos con arritmia ventricular sostenida espontánea asociada con disfunción ventricular grave del ventrículo sistémico siempre que estén recibiendo tratamiento médico apropiado y que tengan una expectativa de vida mayor de un año. (Nivel de evidencia B.)
2. La terapia con CDI en conjunto con el tratamiento médico/quirúrgico es razonable en los pacientes con riesgo alto de MS o arritmias ventriculares sostenidas asociado con afectación genética (canalopatías o miocardiopatías) o con CC. (Nivel de evidencia C.)
3. La terapia con CDI es razonable en pacientes con CC que han presentado síncope de causa inexplicable y/o tienen disfunción ventricular grave y/o arritmias ventriculares sostenidas inducibles en ausencia de una causa reversible y siempre que reciban tratamiento médico-quirúrgico óptimo y tengan una expectativa de vida mayor de un año. (Nivel de evidencia B.)
4. La evaluación hemodinámica mediante cateterismo cardíaco y/o RMN y el EEF es razonable en los pacientes con CC y TV sostenida. (Nivel de evidencia C.)
5. La evaluación hemodinámica invasiva mediante cateterismo cardíaco y/o RMN y el EEF es razonable en pacientes con CC asociadas con síncope de causa inexplicable y/o disfunción ventricular. (Nivel de evidencia B.)
Clase IIb
1. El EEF puede considerarse para los pacientes con CC y duplas ventriculares o TVNS para determinar el riesgo de arritmia ventricular sostenida. (Nivel de evidencia C.)
2. La ablación por catéter puede considerarse en niños y adolescentes con TV monomórfica idiopática repetitiva no sostenida refractaria al tratamiento médico aunque no cause síntomas ni disfunción ventricular. (Nivel de evidencia C.)
3. La ablación por catéter puede considerarse en pacientes con TV/FV refractarios, en quienes la EV desencadenante del evento es siempre de la misma morfología. (Nivel de evidencia C.)
Clase III
1. El tratamiento antiarrítmico no está indicado para los pacientes pediátricos asintomáticos con EV simples, monomórficas, sin cardiopatía demostrable. (Nivel de evidencia C.)
2. La terapia antiarrítmica profiláctica no está indicada para los pacientes asintomáticos con CC con EV simples y aisladas. (Nivel de evidencia C.)
CAPíTULO 9
Otras patologías
Ventrículo izquierdo no compacto
El ventrículo izquierdo no compacto (VINC) es una patología cardíaca infrecuente de origen genético, agrupada dentro de las miocardiopatías no clasificadas, también conocida como miocardiopatía espongiforme (83).
Características
Alteración de la estructura del miocardio ventricular izquierdo con gran trabeculación y formación de recesos intertrabeculares que generan capas de miocardio compacto y no compacto (84). En algunas ocasiones puede asociarse con fístulas coronarias y alteraciones cardíacas congénitas, como anomalía de Ebstein, válvula aórtica bicúspide, transposición corregida de los grandes vasos y defectos del tabique interventricular (85). Se debería a un defecto de formación del miocardio fetal en la etapa intrauterina con persistencia del patrón trabecular fetal, con alteración en la regulación de la proliferación, diferenciación y maduración celular. Se transmitiría principalmente en forma autosómica dominante por alteración de los genes Tafazzin, Alpha- distrobrevin (DNTNA) y los correspondientes a las proteínas sarcoméricas (86,87).
Diagnóstico
Electrocardiografía: muestra en general hallazgos comunes a las miocardiopatías, con trastornos de conducción, imagen de bloqueo de rama y alteraciones primarias de la repolarización.
Ecocardiografía: existen varios criterios diagnósticos; entre ellos, los más utilizados son:
a) En estadios típicos y avanzados se observan dos capas miocárdicas definidas, una epicárdica delgada y otra endocárdica engrosada con numerosas y prominentes trabeculaciones en una relación no compacto/compacto > 2:1 en período de fin de sístole.
b) Flujo Doppler intertrabecular.
c) Malla trabecular prominente en el ápex o en los segmentos inferomedial y lateral (88).
Resonancia magnética nuclear cardíaca: presencia de hipertrabeculación en el ápex, en la cara lateral e inferior del VI con una relación de capas no compacto/compacto > 2,3, medido en período diastólico (89).
Pronóstico
Es de alta morbimortalidad a mediano plazo en pacientes sintomáticos, determinada por su potencial evolución a formas de IC refractaria por disfunción sistólica, stroke con y sin FA y ocurrencia de MS, que es la causa principal de muerte por su asociación con arritmias ventriculares malignas, lo que justificaría la colocación de un CDI. Por lo mencionado, también suele evaluarse para trasplante cardíaco.
Prevención secundaria
Clase I
1. El implante de un CDI está indicado en sobrevivientes de MS por FV o TV sostenida hemodinámicamente inestable en ausencia de causas reversibles. (Nivel de evidencia A.)
Prevención primaria
Clase I
1. El implante de un CDI está indicado en pacientes con cardiopatía estructural y TV espontánea, sea estable o inestable hemodinámicamente. (Nivel de evidencia B.)
Clase IIb
1. El implante un CDI podría estar indicado en pacientes con VINC y antecedentes de síncope, TVNS, Fey £ 35% o historia familiar de MS. (Nivel de evidencia C.)
2. El implante de un CDI podría considerarse en pacientes con VINC asociado con antecedentes familiares de MS. (Nivel de evidencia C.)
Fibrilación ventricular idiopática
En la actualidad, el diagnóstico de fibrilación ventricular idiopática (FVI), también llamada enfermedad eléctrica primaria, es por exclusión. Se trataría de pacientes con FV sin cardiopatía estructural evidenciable por ecocardiografía, sin enfermedad coronaria detectable por angiografía coronaria o ergometría, sin alteraciones electrocardiográficas de la repolarización y resonancia magnética nuclear normal. Se excluyen, por lo tanto, dentro de esta definición a los pacientes portadores de síndrome de QT largo o corto, síndrome de Brugada y TV catecolaminérgica. Se estima que esta enfermedad es responsable de hasta el 5% de los casos de MS (90).
Electrocardiografía: en general, el ECG es morfológicamente normal y con intervalo QTc normal en condiciones basales, con algunas excepciones. En pacientes con antecedentes de síncope y MS se han observado ECG con onda J y repolarización ventricular precoz en cara inferior y lateral, aunque no existen todavía datos concluyentes sobre esta asociación (91,92). En ocasiones se han detectado pacientes con extrasístoles ventriculares con imagen de bloqueo de rama izquierda con eje extremo derecho e intervalo de acoplamiento muy corto como gatillo de inicio de la FV (extrasístole ventricular de la red de Purkinje o del tracto de salida del VD). En general, la forma de presentación más reconocida es de TV polimórfica sostenida o no sostenida que se transforma rápidamente en FV.
Electrofisiología: los intervalos electrofisiológicos intracavitarios (AH y HV) son normales, de la misma manera que los períodos refractarios. La inducción de taquiarritmias polimórficas sostenidas es un marcador de muy mal pronóstico en pacientes que han tenido FVI y en general esta respuesta es abolida por drogas antiarrítmicas de clase Ia. Con protocolos agresivos, la inducibilidad llega a ser positiva en hasta 89% de los casos con FVI, a diferencia de los sujetos normales, en los que puede llegar hasta 9% (93).
Pronóstico y tratamiento: la recurrencia de FVI se estima en alrededor de 22% y de 37% en los dos y cuatro años siguientes al primer episodio, respectivamente (93). Dada la ausencia de cardiopatía estructural, el pronóstico es aceptablemente bueno si los episodios arrítmicos son abolidos o controlados. Las alternativas posibles son: CDI, ablación por radiofrecuencia y fármacos antiarrítmicos (quinidina), pero nunca como monoterapia, sino combinadas. Las comunicaciones existentes sobre esta rara patología son suficientes para realizar la indicación del CDI como prevención secundaria de MS en los sobrevivientes.
La ablación por radiofrecuencia de focos extrasistólicos desencadenantes se ha utilizado con fines curativos y para disminuir la frecuencia de descargas del CDI (94). Ocasionalmente, en pacientes con FVI o TV polimórfica maligna pueden encontrarse arritmias ventriculares originadas en el tracto de salida del VD. La ablación por radiofrecuencia es una alternativa válida para esta entidad (95).
Prevención secundaria
Clase I
1. El implante de un CDI está indicado en pacientes sobrevivientes de MS por FV o TV sostenida hemodinámicamente inestable después de excluir causas reversibles. (Nivel de evidencia A.)
Prevención primaria
Clase I
1. El implante de un CDI está indicado en pacientes con síncope de origen indeterminado, clínicamente relevante, con TV sostenida con compromiso hemodinámico o FV inducibles en el EEF. (Nivel de evidencia B.)
Taquicardia ventricular polimórfica catecolaminérgica
La taquicardia ventricular polimórfica catecolaminérgica (TVPC) se caracteriza por la aparición de TVP inducida por estrés emocional o ejercicio físico, en ausencia de cardiopatía estructural. También es conocida como TVPC familiar por su carácter genético hereditario, aunque puede aparecer como mutación de novo en pacientes sin historia familiar de TVPC. En muchas ocasiones, la taquicardia ventricular polimórfica sostenida sincopal o la FV son el primer síntoma de la enfermedad.
Genética
Se reconocen mutaciones en dos genes como responsables de la TVPC:
1. Ryanodine receptor (RyR2) o canal cardíaco sarcoplasmático liberador de calcio. La activación de este canal con el ejercicio o estimulación betaadrenérgica aumentaría la liberación de calcio intracelular gatillando la aparición de TVP. Su transmisión sería de tipo autosómico dominante ligado al cromosoma 1q42-q43.
2. Calsequestrin2 (CASQ2); esta mutación sería del tipo autosómico recesivo. Produciría una alteración en la dinámica celular de la calsequestrina 2, proteína con actividad de reservorio de calcio intracelular (96,97).
Electrocardiografía
En condiciones basales, el ECG es normal, sin alteraciones de la repolarización y con intervalo QT normal. La complejidad de la arritmia en general guarda relación con los niveles de ejercicio; al inicio del ejercicio aparecen taquicardia sinusal, extrasistolia supraventricular y ventricular frecuente y continúa con TV polimórfica desencadenada por una EV que evoluciona a formas sostenidas o no sostenidas. La presentación como TV bidireccional también es una forma frecuente de encontrar en esta patología.
Estudio electrofisiológico
Los intervalos electrofisiológicos intracavitarios son normales y la inducibilidad de TV o FV es negativa. El mecanismo de la arritmia estaría relacionado con la presencia de circuitos de microrreentrada y postpotenciales tardíos dependientes del calcio y/o variantes del tono autonómico.
Pronóstico
No es posible realizar una estratificación pronóstica adecuada por tratarse de una patología de baja prevalencia; sin embargo, se acepta que en pacientes jóvenes sintomáticos por síncope o TV hemodinámicamente inestable el riesgo de MS sin tratamiento es elevado. En general, el tratamiento con betabloqueantes suele ser efectivo en la mayoría de los pacientes. El marcador de peor pronóstico sería la recurrencia de la taquicardia ventricular en pacientes tratados con betabloqueantes (98).
Tratamiento
- Ejercicio físico
Clase I
1. La restricción de la actividad física es obligatoria.
- Betabloqueantes
Su utilización es efectiva en la prevención de recurrencias arrítmicas y síncope, con algunas excepciones y consideraciones. Es necesaria una adherencia al tratamiento cercana a 100% para evitar el síndrome de supresión de fármaco y la aparición de arritmias ventriculares. No negativiza totalmente la inducción de arritmias supraventriculares o ventriculares con el ejercicio. Es obligatoria su administración en todos los casos con diagnóstico genético a edad temprana.
Clase I
1. Se indica la administración de betabloqueantes en los portadores de las alteraciones genéticas de la TVPC, con diagnóstico a edad temprana y arritmias espontáneas o inducidas por el ejercicio. (Nivel de evidencia C.)
Clase IIa
1. Se indica la administración de betabloqueantes en los portadores asintomáticos de las alteraciones genéticas de la TVPC y con diagnóstico a edad temprana. (Nivel de evidencia C.)
Clase IIb
1. Se indica la administración de betabloqueantes en los portadores asintomáticos de las alteraciones genéticas de la TVPC y con diagnóstico en la edad adulta. (Nivel de evidencia C.)
- Cardiodesfibrilador
Existe acuerdo sobre su utilización en pacientes sintomáticos bajo tratamiento con betabloqueantes, resucitados y en los portadores del gen con historia familiar de MS (99).
Prevención secundaria
Clase l
1. El implante de un CDI y el tratamiento con betabloqueantes está indicado en pacientes con TVPC sobrevivientes de MS con expectativa de vida mayor de un año. (Nivel de evidencia C.)
Prevención primaria
Clase IIa
1. El implante de un CDI es aconsejable en pacientes con TVPC que han tenido síncope o taquicardia ventricular sostenida bajo tratamiento con betabloqueantes. (Nivel de evidencia C.)
Bibliografía
1. Schwartz PJ, Periti M, Melliani A. The long Q-T syndrome. Am Heart J 1975; 89: 378-90.
2. Dessertenne F. Ventricular tachycardia with 2 variable opposing foci. Arch Mal Coeur 1966; 59: 263-7.
3. Acunzo RS. Las taquicardias ventriculares multiformes en pacientes con intervalo QT prolongado. En: Elizari MV, Chiale PA, editores. Arritmias cardíacas. Bases celulares, diagnóstico y tratamiento. Editorial Propulsora Literaria 1998; cap. 20, p. 395-413.
4. Rautaharju PM, Surawicz B, Gettes LS, Bailey JJ, Childers R, Deal BJ, et al. AHA/ACCF/HRS. Recommendations for the standardization and interpretation of the electrocardiogram. Part IV: The ST Segment, T and U Waves, and the QT Interval. J Am Coll Cardiol 2009; 53: 982-91.
5. Jervell A, Lange-Nielsen F. Congenital deaf mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957; 54: 59-68.
6. Lu JT, Kass RS. Recent progress in congenital long QT syndrome. Curr Opin Cardiol 2010; 25: 216-21.
7. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. Guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. ACC/AHA/ ESC Europace 2006; 8: 746-837.
8. Medeiros-Domingo A, Iturralde-Torres P, Ackerman MJ. Clínica y genética en el síndrome de QT largo. Rev Esp Cardiol 2007; 60: 739-52.
9. Shimuzu W. The long QT syndrome: Therapeutic implications of a genetic diagnosis. Cardiovasc Res 2005; 67: 347-56.
10. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long Q-T syndrome: An update. Circulation 1993; 88: 782-4.
11. Imboden M, Swan H, Denjoy I, Van Langen IM, Latinen-Forsblom PJ, Napolitano C, et al. Female predominance and transmission distortion in long QT syndrome. N Engl J Med 2006; 355: 2744-51.
12. Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol 2010; 55: 2745-52.
13. Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V, et al. Prevention of torsade de pointes in hospital settings. AHA/ ACCF. Circulation 2010; 121: 1047-60.
14. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace 2006; 8: 746-837.
15. Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA 3rd, Freedman RA, Gettes LS, et al. ACC/AHA/HRS Guidelines for device-based therapy of cardiac rhythm abnormalities. Circulation 2008; 117: 350-408.
16. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20: 1391-6.
17. Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome. Eur Heart J 2002; 23: 1648-54.
18. Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation 2000; 102: 2509-15.
19. Belhassen B, Viskin S, Fish R, Glick A, Setbon I, Eldar M. Effects of electrophysiologic-guided therapy with Class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fibrillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol 1999; 10: 1301-12.
20. Postema P, Wolpert C, Amin A, Probst V, Borggrefe M, Roden D, et al. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm 2009; 6: 1335-41.
21. Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med 1990; 89: 588-96.
22. Peters S, Peters H, Thierfelder L. Risk stratification of sudden cardiac death and malignant ventricular arrhythmias in right ventricular dysplasia-cardiomyopathy. Int J Cardiol 1999; 71: 243-50.
23. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988; 318: 129-33.
24. Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation 1992; 86: 29-37.
25. Roberts R, Schwartz K. Myocardial diseases. Circulation 2000; 102: 34-9.
26. Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM, Cannom DS, et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol 2009; 54: 609-15.
27. Calkins H, Marcus F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: an update. Curr Cardiol Rep 2008; 10: 367-75.
28. Arbelo E, Josephson ME. Ablation of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol 2010; 21: 473-86.
29. Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003; 108: 3084-91.
30. Gillis AM, Sheldon RS, Wyse DG, Duff HJ, Cassidy MR, Mitchell LB. Clinical and electrophysiologic predictors of ventricular tachyarrhythmia recurrence in patients with implantable cardioverter defibrillators. J Cardiovasc Electrophysiol 2003; 14: 492-8.
31. Hodgkinson KA, Parfrey PS, Bassett AS, Kupprion C, Drenckhahn J, Norman MW, et al. The impact of implantable cardioverter- defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5). J Am Coll Cardiol 2005; 45: 400-8.
32. Pezawas T, Stix G, Kastner J, Schneider B, Wolzt M, Schmidinger H. Ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy: clinical presentation, risk stratification and results of long-term follow-up. Int J Cardiol 2006; 107: 360-8.
33. Roguin A, Bomma CS, Nasir K, Tandri H, Tichnell C, James C, et al. Implantable cardioverter-defibrillators in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2004; 43: 1843-52.
34. Tavernier R, Gevaert S, De SJ, De Clercq A, Rottiers H, Jordaens L, et al. Long term results of cardioverter-defibrillator implantation in patients with right ventricular dysplasia and malignant ventricular tachyarrhythmias. Heart 2001; 85: 53-6.
35. Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation 2004; 109: 1503-8.
36. Piccini JP, Dalal D, Roguin A Bomma C, Cheng A, Prakasa K, et al. Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia. Heart Rhythm 2005; 11: 1188-94.
37. Wichter T, Breithardt G. Implantable cardioverter-defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: a role for genotyping in decision-making? J Am Coll Cardiol 2005; 45: 409-11.
38. Maron BJ, McKenna WJ, Danielson GK, Kappenbergr LJ, Kuhn HJ, Seidman CE, et al. ACC/ESC clinical expert consensus document on hypertrophic cardiomyopathy: a report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines (Committee to Develop an Expert Consensus Document on Hypertrophic Cardiomyopathy). Eur Heart J 2003; 21: 1965-91.
39. Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol 2010; 25: 205-9.
40. Casabé JH, Acunzo R, Fernández A, Gabay J, Galizio N, Hita A, et al. Consenso Argentino de Miocardiopatía Hipertrófica. Rev Argent Cardiol 2009; 77: 151-66.
41. Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA 3rd, Freedman RA, Gettes LS, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac abnormalities. Circulation 2008; 117: 350-408.
42. Moss AJ, Hall WJ, Cannon DS, Daubert JP, Higgins SL, Klein H, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med 1996; 335: 1933-40.
43. Bigger JT. Prophylactic use of implanted cardiac defibrillators in patients at high risk for ventricular arrhythmias after coronary artery bypass graft surgery. Coronary artery bypass graft (CABG) patch trial investigators. N Engl J Med 1997; 337: 1569-75.
44. Buxton AE, Lee KL, Fischer JD, Josephson ME, Prystowsky EN, Hafley H. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med 1999; 341: 1882-90.
45. Buxton AE, Lee KL, DiCarlo L, Gold MR, Greer GS, Prystowsky EN, et al. Electrophysiologic testing to identify patients with coronary artery disease who are at risk for sudden death. N Engl J Med 2000; 342: 1937-45.
46. Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002; 346: 877-83.
47. Hohnloser SH, Kuck KH, Dorian P, Roberts RS, Hampton JR, Hatala R, et al. Prophylactic Use of an Implantable Cardioverter- Defibrillator after Acute Myocardial Infarction. N Engl J Med 2004; 351: 2481-8.
48. Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, et al. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med 2005; 352: 225-37.
49. Myerburg RJ. Implantable cardioverter defibrillators alter myocardial infarction. N Engl J Med 2008; 359: 2245-53.
50. Tung R, Zimetbaum P, Josephson ME. A critical appraisal of implantable cardioverter defibrillators for the prevention of sudden cardiac death. J Am Coll Cardiol 2008; 52: 1111-21.
51. Goldenberg I, Vyas AK, Hall WJ, Moss AJ, Wang H, Zareba W, et al. Risk Stratification for primary implantation of a cardiac defibrillators in patients with ischemic left ventricular dysfunction. J Am Coll Cardiol 2008; 51: 288-96.
52. Zwanziger J, Hall WJ, Dick AW, Zhao H, Muschlin A, Hahn RM, et al. The cost effectiveness of implantable cardioverter defibrillators. Results from MADIT II. J Am Coll Cardiol 2006; 47: 2310-8.
53. Cowie M, Marshall D, Drummond M, Ferko N, Maschio M, Ekman M, et al. Lifetime cost-effectiveness of prophylactic implantation of a cardioverter defibrillator in patients with reduced left ventricular systolic function: results of Markov modelling in an European population. Europace 2009; 11: 716-26.
54. The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med 1997; 337: 1576-83.
55. Kuck KH, Cappato R, Siebels J, Ruppel R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: the Cardiac Arrest Study Hamburg (CASH). Circulation 2000; 102: 748-54.
56. Connolly SJ, Gent M, Roberts RS, Dorian P, Roy D, Sheldon RS, et al. Canadian implantable defibrillator study (CIDS): a randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation 2000; 101: 1297-302.
57. Connolly SJ, Hallstrom AP, Cappato R, Schron EB, Kuck KH, Zipes DP, et al. Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. AVID, CASH and CIDS studies. Antiarrhythmics vs Implantable Defibrillator study. Cardiac Arrest Study Hamburg. Canadian Implantable Defibrillator Study. Eur Heart 2000; 21: 2071-8.
58. Strickberger SA, Hummel JD, Bartlett TG, Frumin HI, Schuger CD, Beau SL, et al. Amiodarone versus implantable cardioverter- defibrillator: randomized trial in patients with nonischemic dilated cardiomyopathy and asymptomatic non sustained ventricular tachycardia - AMIOVIRT. J Am Coll Cardiol 2003; 41: 1707-12.
59. Bänsch D, Antz M, Boczor S, Volkmer M, Tebbenjohanns J, Seidl K, et al. Primary prevention of sudden cardiac death in idiopathic dilated cardiomyopathy: The Cardiomyopathy Trial (CAT). Circulation 2002; 105: 1453-8.
60. Kadish A, Dyer A, Levine J, Quigg R, Estes NA, Anderson KP, et al. Prophylactic Defibrillator Implantation in Patients with Non- ischemic Dilated Cardiomyopathy (DEFINITIVE). N Engl J Med 2004; 350: 2151-8.
61. Consenso de Arritmias Ventriculares. Consejo de Electrocardiografía, Electrofisiología, Arritmias y Marcapasos “Dr. Antonio Batro”. Rev Argent Cardiol 2002; 70(Supl 4).
62. Rassi A Jr. Implantable Cardioverter-Defibrillators in patients with Chagas heart disease: misperceptions, many questions and the urgent need for a randomized clinical trial. J Cardiovasc Electrophysiol 2007; 18: 1241-3.
63. Consenso de Enfermedad de Chagas, Consejo de Enfermedad de Chagas y Miocardiopatías Infecciosas “Dr. Salvador Mazza”. Rev Argent Cardiol 2002; 70(Supl 1): 1-87.
64. Viotti R, Vigliano C, Lococo B, Petti M, Bertocchi G, Álvarez MG, et al. Indicadores clínicos de progresión de la miocarditis chagásica crónica. Rev Esp Cardiol 2005; 58: 1037-44.
65. Cardinalli-Neto A, Greco O, Bestetti R. Automatic Implantable Cardioverter-Defibrillators in Chagas’ Heart Disease Patients with Malignant Ventricular Arrhythmias. Pacing Electrophysiol 2006; 29: 467-70.
66. Cardinelli-Neto A, Bestetti R, Cordeiro J, Rodrigues V. Predictors of all-cause mortality for patients with chronic Chagas’ heart disease receiving implantable cardioverter defibrillator therapy. J Cardiovasc Electrophysiol 2007; 18: 1236-40.
67. Rassi A Jr, Rassi A, Rassi S. Predictors of mortality in chronic Chagas disease. A systematic review of observational studies. Circulation 2007; 115: 1101-8.
68. Dubner S, Valero E, Pesce R, Zuelgaray JG, Mateos JC, Filho SG, et al. A Latin American registry of implantable cardioverter defibrillators: the ICD-LABOR study. Ann Noninvasive Electrocardiol 2005; 10: 420-8.
69. Muratore C, Batista L, Chiale P, Eloy R, Tentori MC, Escudero J, et al. Implantable cardioverter defibrillators and Chagas’ disease results of the ICD Registry Latin America. Europace 2009; 11: 164-8.
70. Murature C, Rabinovich R, Iglesias R, González M, Darú V, Sosa Liprandi A. Implantable cardioverter defibrillators in patients with Chagas’ disease: are they different from patients with coronary disease? Pacing Electrophysiol 1997; 20: 194-7.
71. Rassi A Jr, Rassi A, Little W, Xavier S, Rassi S, Rassi AG, et al. Development and validation of a risk score for predicting death in Chagas’ heart disease. N Engl J Med 2006; 355: 799-808.
72. Wren C, O’Sullivan JJ, Wright C. Sudden death in children and adolescents. Heart 2000; 83: 410-3.
73. Silka MJ, Kron J, Dunnigan A, Dick M 2nd. Sudden cardiac death and the use of implantable cardioverter-defibrillators in pediatric patients. The Pediatric Electrophysiology Society. Circulation 1993; 87: 800-7.
74. Strain JE, Grose RM, Factor SM, Fisher JD. Results of endomyocardial biopsy in patients with spontaneous ventricular tachycardia but without apparent structural heart disease. Circulation 1983; 68: 1171-81.
75. Ryujin Y, Arakaki Y, Takahashi O, Kamiya T. Ventricular arrhythmias in children: the validity of exercise stress tests for their diagnosis and management. Jpn Circ J 1984; 48: 1393-8.
76. Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002; 106: 69-74.
77. Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 1998; 338: 1709-14.
78. Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomized trials. Lancet 1997; 350: 1417-24.
79. Dubin AM, Berul CI, Bevilacqua LM, Cillins KK, Etherigde SP, Fenrich AL, et al. The use of implantable cardioverter-defibrillators in pediatric patients awaiting heart transplantation. J Card Fail 2003; 9: 375-9.
80. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med 2003; 348: 1866-74.
81. McKenna WJ, Behr ER. Hypertrophic cardiomyopathy: management, risk stratification, and prevention of sudden death. Heart 2002; 87: 169-76.
82. Alexander ME, Cecchin F, Walsh EP, Triedman JK, Bevilacqua LM, Berul CI. Implications of implantable cardioverter defibrillator therapy in congenital heart disease and pediatrics. J Cardiovasc Electrophysiol 2004; 15: 72-6.
83. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from European Society of Cardiology working group on myocardial and pericardial diseases. Eur Heart J 2008; 29: 270-6.
84. Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, et al. Clinical characterization of left ventricular non- compaction in children: a relatively common form of cardiomyopathy. Circulation 2003; 108: 2672-8.
85. Lilje C, Rázek V, Joyce JJ, Rau T, Finckh BF, Weiss F, et al. Complications of non-compaction of the left ventricular myocardium in a paediatric population: a prospective study. Eur Heart J 2006; 27: 1855-60.
86. Henderson DJ, Anderson RH. The development and structure of the ventricles in the human heart. Pediatr Cardiol 2009; 30: 588-96.
87. Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008; 117: 2893-901.
88. Jenni R, Oechnlin E, Schneider J, Jost CA, Kaufman PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001; 86: 666-71.
89. Petersen SE, Selvanayagam JB, Wiesmann F, Robson MD, Francis JM, Anderson RH, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol 2005; 46: 101-5.
90. Consensus Statement of the Joint Steering Committees of the Unexplained Cardiac Arrest Registry of Europe and of the Idiopathic Ventricular Fibrillation Registry of the United States: survivors of out-of-hospital cardiac arrest with apparently normal heart. Need for definition and standardized clinical evaluation. Circulation 1997; 95: 265-72.
91. Haissaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008; 358: 2016-23.
92. Tikkanen JT, Anttonen O, Junttila MJ, Aro AL, Kerola T, Rissanen HA, et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med 2009; 361: 2529-37.
93. Morady F, DiCarlo L, Baerman J, de Buitleir M. Comparison of coupling intervals that induce clinical and nonclinical forms of ventricular tachycardia during programmed stimulation. Am J Cardiol 1986; 57: 1269-73.
94. Haissaguerre M, Shoda M, Jais P, Nogami A, Shah DC, Kautzner J, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation 2002; 106: 962-7.
95. Noda T, Shimizu W, Taguchi A, Aiba T, Satomi K, Suyama K, et al. Malignant entity of idiopathic ventricular fibrillation and polymorphic ventricular tachycardia initiated by premature extrasystoles originating from the right ventricular outflow tract. J Am Coll Cardiol 2005; 46: 1288-94.
96. Lahat H, Pras E, Eldar M. RYR2 and CASQ2 mutations in patients suffering from catecholaminergic polymorphic ventricular tachycardia. Circulation 2003; 107: 29.
97. Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001; 103: 485-90.
98. Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002; 106: 69-74.
99. Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-Year follow-up of 21 patients. Circulation 1995; 91: 1512-9.


{kind=link}
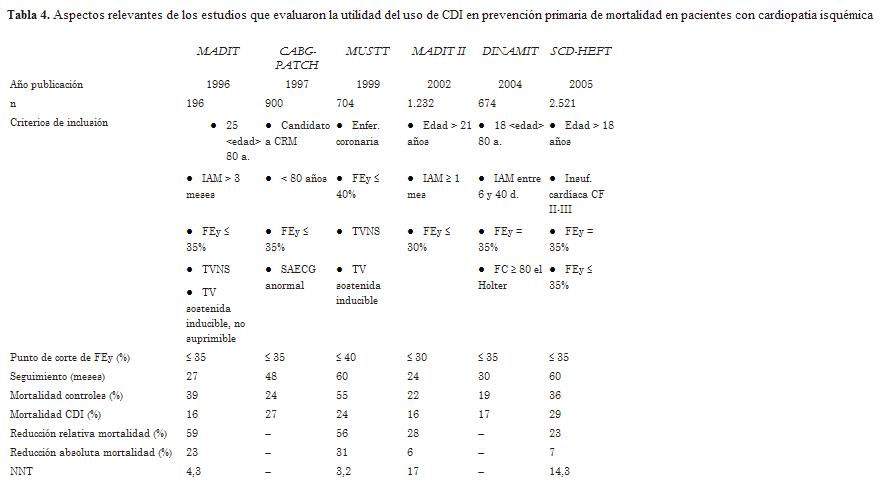{kind=link}
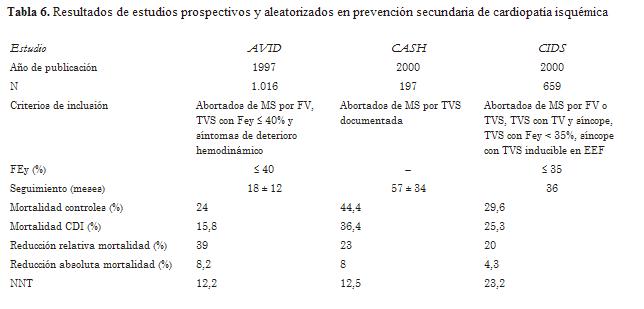{kind=link}