










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Uruguaya de Cardiología
versión On-line ISSN 1688-0420
Rev.Urug.Cardiol. vol.26 no.1 Montevideo mar. 2011
REGISTRO URUGUAYO
Miocardiopatía hipertrófica
Aspectos conceptuales de la enfermedad y fundamentos del Registro Uruguayo
de Miocardiopatía Hipertrófica
Dres. Jorge Estigarribia Passaro, Inés Vidal Cortinas, Álvaro Báez, Juan Luis Vidal
Co-Coordinadores del Registro Uruguayo de Miocardiopatía Hipertrófica
Correspondencia: Dr. Jorge Estigarribia, correo eléctronico: jorgeestigarribia@movinet.com.uy
Recibido mayo 9, 2011; aceptado mayo 11, 2011
RESUMEN
La miocardiopatía hipertrófica (MCH) es una miocardiopatía primaria, de transmisión genética, autosómica dominante, con una prevalencia en la población general adulta de 1 en 500, caracterizada por una hipertrofia parietal fundamentalmente del ventrículo izquierdo, en ausencia de una enfermedad cardíaca o sistémica que pueda explicarla. A pesar de no ser infrecuente, es una enfermedad subdiagnosticada y de manejo clínico poco sistematizado en la práctica.
El ecocardiograma posee la llave del diagnóstico, y la resonancia nuclear magnética ha realizado aportes recientes en la definición de sus variedades morfológicas.
Tiene un patrón histológico de desorganización arquitectural de las fibras miocárdicas, que son hipertróficas y mal alineadas. Frecuentemente la hipertrofia es asimétrica y puede generar un gradiente dinámico por obstrucción del tracto de salida del ventrículo izquierdo en el cual participa un movimiento anómalo de la válvula mitral (movimiento anterior sistólico; MAS), que habitualmente determina también una insuficiencia valvular.
Los síntomas más frecuentes son la disnea, el dolor anginoso, el síncope y la muerte súbita (MS). Es la primera causa de MS en personas jóvenes y atletas de competición, y un aspecto capital de su manejo clínico es la estratificación del riesgo de esta trágica complicación para decidir el implante oportuno de un cardiodesfibrilador automático.
A través de la Sociedad Uruguaya de Cardiología se ha instrumentado un Registro (RUMHI) de portadores de esta patología que pretende mejorar el conocimiento de la entidad, obtener una aproximación a su frecuencia y conocer su evolución, pronóstico y forma de tratamiento en Uruguay.
Palabras clave: CARDIOMIOPATÍA HIPERTRÓFICA
SUMMARY
Hypertrophic cardiomyopathy is a primary cardiomyopathy, inherited as an autosomal dominant threat, with prevalence in the general adult population of 1 in 500, primarily characterized by parietal hypertrophy of the left ventricle, in the absence of a cardiac or systemic disease that could explain it. Despite not being uncommon, it is an underdiagnosed disease and its clinical management is little systematized in practice.
Echocardiography has the key to the diagnosis, and magnetic resonance imaging has made recent contributions in the definition of its morphological varieties.
It has a histological pattern of architectural disarray of myocardial fibers, which are hypertrophic and poorly aligned. Hypertrophy is often asymmetric and can generate a dynamic gradient by obstruction of the outflow tract of the left ventricle which involved an anomalous motion of the mitral valve (systolic anterior movement, SAM), which usually also determines a valvular insufficiency.
The most common symptoms are breathlessness, chest pain, syncope and sudden death (SD). It is the first cause of SD in young people and athletes in competition, and a capital aspect of its clinical management is the stratification of the risk of this tragic complication to decide to implant an automatic cardioverter-defibrillator timely.
Through Sociedad Uruguaya de Cardiología, a Registry of carriers of this disease has been implemented (RUMHI) which aims to improve the knowledge of the entity, obtain an approximation to the frequency and know its evolution, prognosis and treatment in Uruguay.
Key words: CARDIOMIOPATHY, HYPERTROPHIC
INTRODUCCIóN
Las miocardiopatías constituyen un grupo heterogéneo de enfermedades cuya clasificación y nomenclatura han permanecido bajo un manto de relativa incertidumbre y confusión. En la actualidad ha logrado aceptación una reciente propuesta de clasificación de la American Heart Association. En esta se las presenta como “una variedad de enfermedades del miocardio que determinan una disfunción mecánica y/o eléctrica, exhiben habitualmente (pero no invariablemente) hipertrofia o dilatación ventricular inapropiadas, y son debidas a una diversidad de causas, que frecuentemente son de orden genético” (1).
En este contexto, la miocardiopatía hipertrófica (MCH) constituye una miocardiopatía primaria, es decir que no se trata de una expresión más de un desorden sistémico o generalizado (como ocurre, por ejemplo, en la amiloidosis), sino que la anormalidad se encuentra confinada al corazón, constituyendo toda la enfermedad. Se caracteriza por una hipertrofia parietal de uno de los ventrículos (habitualmente el izquierdo) o de ambos, de distribución típicamente asimétrica, sin dilatación y en ausencia de una enfermedad cardíaca o sistémica capaz de explicarla (2). Por tanto, la existencia de condiciones de sobrecarga ventricular de suficiente magnitud y persistencia como para generar una hipertrofia adaptativa o secundaria (como la hipertensión arterial y la estenosis aórtica severas), descartan el diagnóstico de la enfermedad.
Histopatológicamente el patrón distintivo es la desorganización arquitectural de las fibras miocárdicas, que son hipertróficas, con variaciones de su tamaño y mal alineadas. Típicamente esta disposición se presenta en más de 5% de la masa ventricular. El desarreglo de miocitos se acompaña en general de un aumento del tejido fibroso intersticial y un engrosamiento de la capa media de las arteriolas intramiocárdicas a expensas de una reducción de su luz (3,4).
La distribución de la hipertrofia en la pared ventricular es variable, aunque en su forma más típica predomina en el sector anterior del septum interventricular basal (figura 1), dando fundamento a la denominación con que Teare hizo conocer la entidad en su publicación original: hipertrofia septal asimétrica (3). Sin embargo, este no es un requisito excluyente, y cualquier patrón de distribución es posible, como lo han demostrado las revisiones ecocardiográficas (5) y especialmente los más recientes estudios con resonancia nuclear magnética (6,7).
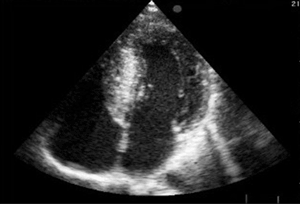
Figura 1. Ecocardiograma bidimensional. Enfoque apical de cuatro cámaras. Severa hipertrofia asimétrica a predominio del septum basal y medio en un paciente no hipertenso.
La hipertrofia parietal casi nunca está presente al momento de nacimiento, y si bien la oportunidad de su adquisición en el curso de la vida del sujeto es variable, con frecuencia ocurre durante la adolescencia, etapa de desarrollo muscular más activo (8). Esto determina que exista un período en el cual el paciente es portador de la enfermedad (por poseer el defecto genético) pero no es aún detectable mediante estudios de imagen. Este período de enfermedad morfológicamente oculta o latente se ha denominado miocardiopatía hipertrófica sin hipertrofia (9) o etapa preclínica (10).
Múltiples denominaciones posteriores, actualmente en desuso, han centrado el énfasis en otros aspectos de la enfermedad, especialmente en una característica que hoy sabemos que es frecuente pero no constante: la obstrucción del tracto de salida del ventrículo izquierdo* (TSVI).
Esta obstrucción se origina tanto en la protrusión del septum hipertrófico como en un movimiento sistólico anterior (MAS) de la valva anterior de la mitral que puede llegar al contacto mitroseptal, generando un gradiente de presión que típicamente es dinámico, es decir que varía con las condiciones de carga ventricular y el estado contráctil del miocardio (11-13). La obstrucción entonces puede estar siempre ausente, puede estar presente en condiciones basales, o únicamente manifestarse con maniobras o fármacos que disminuyen la precarga o la poscarga y/o incrementan la contractilidad miocárdica (obstrucción latente).
Cuando existe obstrucción del TSVI, el MAS de la valva anterior de la mitral genera un defecto de coaptación sistólica que lleva a la insuficiencia valvular, que también es dinámica y de magnitud paralela a la severidad del gradiente de la vía de salida (14). En la génesis de esta insuficiencia mitral no resulta necesaria una anomalía estructural de la válvula, pero no es infrecuente la existencia de valvas engrosadas, elongadas o incluso con franco prolapso, o bien anomalías de posición o inserción del músculo papilar anterior que favorecen tanto la obstrucción como la regurgitación valvular (15) y que deben ser tenidas en cuenta en caso de indicación quirúrgica (16).
De la fisiopatología de la MCH, además de la frecuente obstrucción del TSVI y la insuficiencia mitral, deben destacarse la casi siempre presente disfunción diastólica, producto tanto de la mayor rigidez pasiva de la cámara (por la geometría ventricular y la fibrosis miocárdica) como del déficit de relajación activa (17), y la isquemia miocárdica, que reconoce su origen en una microcirculación anatómica y funcionalmente anormal, una desproporción entre el lecho capilar y la incrementada masa muscular, la mayor presión intramural y el aumento del consumo de oxígeno (18-20). Esta isquemia microvascular tiene como consecuencias, en primer lugar, la posibilidad de angina con coronarias epicárdicas normales, y,k por otra parte, una sustitución fibrosa progresiva de los miocitos, que en una proporción mínima de pacientes puede causar un cambio radical en el patrón de remodelación del ventrículo izquierdo, con adelgazamiento parietal, dilatación de la cavidad, deterioro marcado de la función sistólica y desaparición de la obstrucción si estaba presente (21-23). En esta infrecuente fase terminal de la MCH el aspecto macroscópico del ventrículo es indiferenciable de una miocardiopatía dilatada de origen isquémico.
La desorganización histoarquitectural de los miocitos, la fibrosis, la disfunción diastólica, la hipertonía adrenérgica, la insuficiencia mitral y la isquemia miocárdica favorecen la génesis de un amplio espectro de arritmias cardíacas, que van desde la extrasistolía de cualquier origen pasando por la fibrilación auricular –que frecuentemente es causa de inicio o agravación de síntomas e incrementa abruptamente el riesgo de accidente vascular encefálico (AVE) embólico– hasta la temida fibrilación ventricular con muerte súbita (24-27).
Desde hace algo más de dos décadas se reconoce que la causa de la MCH es genética, constituyendo la primera enfermedad cardiovascular en la que se demostró este origen (28), y a la vez la más frecuente. Su modalidad de herencia es mendeliana, autosómica dominante y con variable pero generalmente fuerte penetrancia (29), con lo cual la padecen ambos sexos y habitualmente hay afectados en todas las generaciones en una familia que porta la enfermedad. Mayoritariamente los casos son familiares, producto de herencia paterna o materna, pero en un porcentaje importante son esporádicos, consecuencia de una mutación “de novo”, a partir de la cual la condición es transmisible a la descendencia (30). Se han identificado hasta el presente más de 500 mutaciones posibles en cualquiera de los 13 genes primarios que codifican proteínas del sarcómero cardíaco, y recientemente se han incorporado mutaciones en genes que regulan el metabolismo cardíaco y la homeostasis del calcio, y determinan fenotipos similares, que se conocen como fenocopias (31). Las posibles variaciones morfológicas y clínicas de la enfermedad no responden a una correlación genotipo/fenotipo definida, sino a la existencia de genes secundarios o moduladores y a la influencia ambiental (32). De este modo, es común que en una misma familia existan individuos con distintos patrones de afectación del miocardio, edad de expresión fenotípica, manifestaciones clínicas y riesgo de muerte súbita, a pesar de compartir la misma mutación primaria.
No obstante su reputación como enfermedad rara, en rigor no le es aplicable esta etiqueta, ya que relevamientos ecocardiográficos realizados en diferentes medios geográficos y étnicos han mostrado una prevalencia similar: 1 caso cada 500 individuos en la población general adulta (33-37). De presentarse con la misma frecuencia en nuestro país, puede estimarse la existencia de unos 6.800 pacientes portadores de la enfermedad.
Debe reconocerse que la mayoría de los afectados cursan con síntomas mínimos o ausentes durante la mayor parte de su existencia, y que en un gran porcentaje la condición no representa un menoscabo de la calidad ni la expectativa vital (38). Estos individuos no son detectables en los ámbitos de atención médica, aunque puede llegarse a ellos a través de relevamientos familiares (mediante ecocardiograma o investigación genética) a punto de partida de un caso índice.
Por otra parte, en algunos pacientes la disnea de esfuerzo, el dolor precordial anginoso, las palpitaciones y el síncope o presíncope determinan un deterioro funcional que motiva la consulta y que ocasionalmente resulta invalidante. Si estos síntomas coexisten con obstrucción significativa del TSVI (gradiente mayor de 50 mm Hg) y no responden al tratamiento máximo con betabloqueantes o calcioantagonistas o ambos (39,40), debe recurrirse a procedimientos invasivos como la miectomía septal quirúrgica, la ablación septal percutánea con alcohol o el hoy menos utilizado implante de marcapaso definitivo (41). Es destacable que la miectomía septal operatoria es capaz de lograr no solo un pronunciado alivio del gradiente y de los síntomas, sino también un significativo descenso de la mortalidad por todas las causas y de la muerte súbita en particular, obteniendo una expectativa vital similar a la población general (42). El efecto sobre la muerte súbita parece confirmarse al haberse demostrado una reducción de la incidencia de descargas apropiadas en pacientes portadores de cardiodesfibrilador implantable (43).
Varias complicaciones pueden modificar brusca o gradualmente el curso de la enfermedad. Entre ellas se cuentan la fibrilación auricular (24) y una posible devastadora consecuencia, el AVE embólico (44), la endocarditis infecciosa (45), la evolución progresiva a la fase final dilatada (46) y la muerte súbita por fibrilación ventricular (26).
Esta última reconoce ciertos factores de riesgo (tabla 1) cuyo valor predictivo positivo es limitado (47-50), lo que en prevención primaria ha conducido a criterios disímiles en la decisión de implantar un cardiodesfibrilador automático, la única medida que se ha mostrado eficaz (51,52). Destacamos, sin embargo, que la completa ausencia de estos factores se relaciona con una probabilidad de muerte súbita menor a 1% anual (47) y que en pacientes que experimentaron muerte súbita o taquicardia ventricular sostenida espontánea (prevención secundaria) hay acuerdo universal en el implante del dispositivo (2,50). Otro aspecto notable consiste en que la MCH es la primera causa de muerte súbita en personas jóvenes, especialmente en atletas de competición (53-55), y que la posibilidad de esta trágica complicación no guarda relación directa con el estatus sintomático, por lo que no es raro que se presente como primera manifestación en adolescentes o jóvenes con aparente buen estado de salud (56,57). Esto enfatiza la necesidad de llegar al mayor número posible de portadores y realizar una adecuada estratificación de riesgo que brinde una oportunidad en la prevención primaria de la muerte súbita.
El ECG raramente es normal en la MCH (58), aunque éste puede constituir un subgrupo con enfermedad menos severa y de mejor pronóstico (59). Si bien no describiremos aquí sus posibles alteraciones, que pueden resultar muy orientadoras del diagnóstico (60-63), es interesante destacar que sus cambios pueden preceder en el tiempo al desarrollo de la hipertrofia parietal (64).
El ecocardiograma Doppler constituye la herramienta fundamental en el diagnóstico y en la búsqueda de nuevos casos, al permitir caracterizar la magnitud y el patrón de la hipertrofia, detectar un gradiente dinámico en el TSVI y/o una insuficiencia mitral con o sin anomalías estructurales de la válvula y descartar una causa fija de obstrucción del ventrículo (5). El hallazgo de una hipertrofia masiva (espesor parietal máximo de al menos 30 mm) constituye un factor de riesgo mayor de muerte súbita, y la detección de obstrucción dinámica (gradiente pico del TSVI igual o mayor de 30 mm Hg) representa un factor menor (2,50,60). Las nuevas técnicas con Doppler tisular, strain rate, speckle tracking, eco contraste y eco 3D prometen valiosos aportes en la identificación de la enfermedad en fase preclínica y una mejoría en la seguridad diagnóstica, la estratificación del riesgo y la monitorización del tratamiento (65).
La resonancia nuclear magnética (RNM) ha adquirido en época reciente un valor destacado, especialmente para la caracterización morfológica de la patología en base a su perspectiva tridimensional, en su diferenciación de otras entidades y en la estratificación del riesgo de muerte súbita y no súbita utilizando la técnica de realce tardío de contraste con gadolinio (66-70).
Los estudios de esfuerzo, particularmente el eco estrés con ejercicio físico, tienen utilidad para detectar obstrucción latente (71) y un comportamiento anómalo de la presión arterial intra o postesfuerzo, que adquieren valor como factores de riesgo de muerte súbita (72,73).
El registro electrocardiográfico ambulatorio de Holter aporta una herramienta valiosa para la estratificación del riesgo de muerte súbita y su prevención a través de la detección de corridas de taquicardia ventricular no sostenida (TVNS), que han demostrado particularmente un alto valor predictivo negativo (27,74).
Finalmente, el estudio genético, no disponible en la rutina por su complejidad y costo, constituye el patrón de oro en la detección de portadores de la enfermedad, sobre todo entre familiares de pacientes ya diagnosticados y para la definición diagnóstica en casos fenotípicamente dudosos (75,76). También podría jugar un papel en la evaluación del pronóstico, aunque este punto es motivo de controversia (31,77,78).
La tabla 2 resume los aspectos relevantes de la enfermedad.
POR QUé UN REGISTRO URUGUAYO DE MIOCARDIOPATíA HIPERTRóFICA
En Uruguay desconocemos la frecuencia y la historia natural de la mayoría de las enfermedades cardiovasculares, lo que nos obliga a recurrir, para aproximarnos a esa información, a fuentes de otros países. Un grupo de colegas de la Sociedad Uruguaya de Cardiología (SUC) decidimos intentar un aporte en este sentido, comenzando con la MCH. Elegimos esta patología porque de acuerdo a proyecciones basadas en su prevalencia en otros medios, el número estimado de afectados hace que el proyecto sea factible, porque creemos que en nuestro medio existe un profundo desconocimiento de su epidemiología y porque donde se la ha estudiado, constituye la primera causa de muerte súbita entre los jóvenes. También porque parece erróneamente percibida por muchos colegas como una entidad rara.
Las enfermedades consideradas raras despiertan escasa atención en la comunidad médica y tienen un déficit de diagnóstico en frecuencia y oportunidad, y una subutilización de recursos terapéuticos probadamente eficaces (79).
La MCH, sin embargo, no es una enfermedad rara, aún empleando las definiciones menos exigentes de la Unión Europea (prevalencia menor a 1/2.000 sujetos de la población general) y de Estados Unidos (prevalencia menor a 1/1.500). Comparativamente, se presenta con una frecuencia entre 10 y 50 veces mayor que otras enfermedades que originan un alto interés médico e implican riesgo de muerte brusca, como el síndrome de Marfan, el síndrome de Brugada y el síndrome de QT largo congénito (80).
No obstante, su complejidad genética y fisiopatológica, su variada expresión anatómica y clínica, su rareza percibida y la prevalencia dominante de causas de hipertrofia adaptativa por sobrecarga, en ocasiones superpuestas a la MCH, han condicionado, entre muchos colegas, cierta incomodidad con la enfermedad.
Los objetivos del RUMHI apuntan a mejorar el conocimiento de las presentaciones clínicas y anatómicas de esta entidad, obtener una aproximación a su frecuencia en Uruguay, arrojar luz sobre su evolución y pronóstico –en especial sobre la incidencia de muerte súbita– y conocer la forma en que se estudia y se trata en el país. Existe la fuerte convicción entre los integrantes del proyecto de que la difusión de la información que se obtenga contribuirá a mejorar el diagnóstico y el manejo clínico de esta elusiva enfermedad, incluyendo una más eficaz prevención de la muerte súbita.
ALGUNOS PUNTOS CENTRALES DEL REGISTRO (TABLA 3)
ASPECTOS éTICOS
A pesar de que se trata de un registro nominativo por la necesidad de evitar duplicación de la información, el manejo de los datos personales es estrictamente confidencial, asignando al paciente un número que queda separado de sus datos filiatorios, a los cuales solo podrán acceder los coordinadores del proyecto. Es condición previa a la inclusión del paciente, la obtención de su firma en un consentimiento informado donde se consagra este derecho, entre otros. A su vez, el protocolo del registro recibió la aprobación del Comité de Ética de la SUC y de la Comisión Nacional de Ética en Investigación del Ministerio de Salud Pública. Como todo registro, el RUMHI se limita a la colección sistemática de datos demográficos y de condiciones de salud a ser incorporados a una base de datos definida para un propósito de investigación preespecificado. Por tanto, está exento de todo papel asistencial o educativo en relación directa con el paciente, que le corresponde exclusivamente al profesional a cargo.
Se trata de un registro de participación voluntaria, con lo cual el cumplimiento de sus objetivos depende del interés y la colaboración del cuerpo médico a través de la incorporación de sus pacientes con diagnóstico presuntivo o confirmado de MCH.
CRITERIOS DE INGRESO
El requisito esencial para el ingreso de un paciente es la comprobación (generalmente ecocardiográfica) de una hipertrofia de la pared ventricular de al menos 15 mm en cualquier sector, para la cual no exista una causa cardíaca o sistémica razonable. Esto incluye situaciones en las cuales resulta evidente una desproporción entre la magnitud de la hipertrofia y la entidad de la posible causa, como, por ejemplo, un espesor parietal de 20 mm en un paciente portador de una hipertensión arterial leve. Debe remarcarse que no se requiere ningún patrón particular de distribución de la hipertrofia, y tanto la obstrucción del TSVI como la insuficiencia mitral constituyen datos contingentes. Los pacientes que han sido sometidos a un procedimiento terapéutico invasivo están en condiciones de ser incluidos, y, de hecho, constituyen un grupo de alta certeza diagnóstica. En edad pediátrica (menores de 15 años) el criterio de entrada es un espesor mayor a 2 desvíos estándar de la media obtenida de tablas confeccionadas en base a la talla o al peso corporal.
CóMO INGRESAR A LOS PACIENTES
Debe completarse un formulario de ingreso de una sola faz y muy sencillo de llenar, el cual se encuentra en la página web del registro ( http://www.rumhi.com). Esto se puede realizar directamente “on line” o bajando una versión en pdf para imprimir y llenar, pudiéndose hacer llegar luego a la SUC personalmente o por correo.
asimismo, se completará y enviará un formulario de ecocardiografía, también disponible en las dos formas, con los datos del estudio. Se solicita el envío de imágenes del ecocardiograma Doppler que estableció el diagnóstico, para ser evaluadas por el grupo RUMHI; puede tratarse de videos en formato AVI o MPG (con un “loop” es suficiente) o simplemente la fotografía que a criterio del colega documente mejor la enfermedad, en formato JPG o GIF. Estas imágenes se “suben” directamente a la página del Registro luego de completar el formulario de ingreso.
ENTIDADES AUSPICIANTES
El RUMHI cuenta con el auspicio de la Cátedra de Cardiología, la Facultad de Medicina y la Comisión Honoraria para la Salud Cardiovascular.
Los coordinadores de RUMHI se encuentran a disposición de todos los colegas que deseen plantear dudas o realizar sugerencias (existe un espacio de contacto en la página del RUMHI), y agradecen toda la colaboración que puedan brindar al proyecto mediante el aporte de los casos de que disponen.
BIBLIOGRAFíA
1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies. An American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council of Epidemiology and Prevention. Circulation 2006; 113: 1807-16.
2. Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology Clinical expert consensus document on hypertrophic cardiomyopathy. Eur Heart J 2003; 24: 1965-91.
3. Teare RD. Asymmetrical hypertrophy of the heart in young adults. Br Heart J 1958; 20: 1-8.
4. Almaas BM, Amlie JP. Histopathological changes ad clinical implications in patients with hypertrophic cardiomyopathy. European Cardiology 2010; 6 (2): 88-90.
5. Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol 1995; 26: 1699-708.
6. Syed IS, Ommen SR, Breen JF, Tajik AJ. Hypertrophic cardiomyopathy: Identification of morphological subtypes by echocardiography and cardiac magnetic resonance imaging. J Am Coll Cardiol Img. 2008; 1 (3): 377-9.
7. 7) Maron MS, Maron BJ, Harrigan C, Buros J, Gibson CM, Olivotto I, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol 2009; 54: 220-8.
8. Maron BJ, Spirito P, Wesley I, Arce J. Development in and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med 1986, 315: 610-4.
9. McKenna WJ, Stewart JT, Nihoyannopoulos P, McGinty F, Davies MJ. Hypertrophic cardiomyopathy without hypertrophy: two families with myocardial disarray in absence of increased myocardial mass. Br Heart J 1990; 63: 287-90.
10. Maron BJ, Ho CY. Hypertrophic cardiomyopathy without hypertrophy. An emerging pre-clinical subgroup composed of genetically affected family members. (Edit.). J Am Coll Cardiol Img 2009, 2: 65-8.
11. Yoerger DM, Weyman AE. Hypertrophic obstructive cardiomyopathy: Mechanism of obstruction and response to therapy. Rev Cardiovasc Med 2003; 4 (4): 199-215.
12. Sherrid MV, Gunsburg DZ, Moldenhauer S, Pearle G. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2000; 36: 1344-54.
13. Maron BJ, Maron MS, Wigle ED, Braunwald E. The 50-year history, controversy and clinical implications of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 2009; 54: 191-200.
14. Yu EH, Omran AS, Wigle ED, Williams WG, Siu SC, Rakowski H. Mitral regurgitation in hypertrophic cardiomyopathy: relationship with obstruction and relief wit myectomy. J Am Coll Cardiol 2000; 36: 2219-25.
15. Reis RL, Bolton MR, King JF, Pugh DM, Dunn MI, Mason DT. Antero-superior displacement of papillary muscle producing obstruction and mitral regurgitation in idiopathic hypertrophic subaortic stenosis. Circulation 1974; 50: II-181-8.
16. Balaram SK, Sherrid MV, Derose JJ Jr, Hillel Z, Winson G, Swistel DG. Beyond extended myectomy for hypertrophic cardiomyopathy: The Resection-Plication-Release (RPR) repair. Ann Thorac Surg 2005: 80: 217-23.
17. Wigle ED, Rakowski H, Kimball BP, Williams WG. Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation 1995; 92: 1680-92.
18. Cannon RO 3rd, Rosing DR, Maron BJ, Leon MB, Bonow RO, Watson RM, et al. Myocardial ischemia in patients with hypertrophic cardiomyopathy: contribution of inadequate vasodilator reserve and elevated left ventricular filling pressures. Circulation 1985; 71: 234-43.
19. Knaapen P, Germans T, Camici PG, Rimoldi OE, ten Cate FJ, ten Berg JM, et al. Determinants of coronary microvascular dysfunction in symptomatic hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 2008; 294: H986-H993.
20. Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, Udelson JE, et al. The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol 2009; 54: 866-75.
21. Thaman R, Gimeno JR, Murphy RT, Kubo T, Sachdev B, Mogensen J, et al. Prevalence and clinical significance of systolic impairment in hypertrophic cardiomyopathy. Heart 2005; 91: 920-5.
22. Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F, et al. Relevance of coronary microvascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J Am Coll Cardiol 2006; 47: 1043-8.
23. Sotgia B, Sciagrà R, Olivotto I, Casolo G, Rega L, Betti I, et al. Spatial relationship between coronary microvascular dysfunction and delayed contrast enhancement in patients with hypertrophic cardiomyopathy. J Nucl Med 2008; 49: 1090-6.
24. Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104: 2517-24.
25. Higashikawa M, Nakamura I, Yoshida M, Kinoshita M. Incidence of ischemic strokes in hypertrophic cardiomyopathy is markedly increased if complicated by atrial fibrillation. Jpn Circ J 1997; 61: 673-81.
26. McKenna WJ, Deanfield JE. Hypertrophic cardiomyopathy: an important cause of sudden death. Arch Dis Child 1984; 59: 971-5.
27. Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: An independent marker of sudden death risk in young patients. J Am Coll Cardiol 2003; 42: 873-9.
28. Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 1989; 321: 1372-8.
29. Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy. From bench to the clinics. J Cardiovasc Electrophysiol 2008; 19: 104-110.
30. Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol 2010; 25: 205-9.
31 . Ho CY. Controversies in cardiovascular medicine. Genetics and clinical destiny: Improving care in hypertrophic cardiomyopathy. Circulation 2010; 122: 2430-40.
32. McKenna W, Coccolo F, Elliot PM. Genes and disease expression in hypertrophic cardiomyopathy. Commentary. Lancet 1998; 352: 1162-3.
33. Hada Y, Sakamoto T, Amano K, Yamaguchi T, Takenaka K, Takahashi H, et al. Prevalence of hypertrophic cardiomyopathy in a population of adults Japanese workers as detected by echocardiographic screening. Am J Cardiol 1987; 59: 183-4.
34. 34) Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995; 92: 785-9.
35. Maron BJ, Mathenge R, Casey SA, Poliac LC, Longe TF. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J Am Coll Cardiol 1999; 33: 1590-5.
36. Maron BJ, Spirito P, Roman MJ. Evidence that hypertrophic cardiomyopathy is a common genetic cardiovascular disease: prevalence in a community-based population of middle-aged and elderly American Indians (abstract). Circulation 2003; 108: IV-664.
37. Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: A population-based echocardiographic analysis of 8080 adults. Am J Med 2004; 116: 14-8.
38. Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK, Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA 1999; 281: 650-5.
39. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation 2008; 117: 429-39.
40. Sherrid MV, Pearle G, Gunsburg DZ. Mechanism of benefit of negative inotropes in obstructive hypertrophic cardiomyopathy. Circulation 1998; 97: 41-7.
41. ten Berg J, Steggerda RC, Siebelink HM. The patient with hypertrophic cardiomyopathy. Heart 2010; 96: 1764-72.
42. Ommen SR, Maron BJ, Olivotto I, Maron MS, Cecchi F, Betocchi S, et al. Long term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46: 470-6.
43. McLeod CJ, Ommen SR, Ackerman MJ, Weivoda PL, Shen WK, Dearani JA, et al. Surgical septal myectomy decreases de risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28: 2583-8.
44. Maron BJ, Olivotto I, Bellone P, Conte MR, Cecchi F, Flygenring BP, et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39: 301-7.
45. Spirito P, Rapezzi C, Bellone P, Betocchi S, Autore C, Conte MR, et al. Infective endocarditis in hypertrophic cardiomyopathy. Prevalence, incidence and indications for antibiotic prophylaxis. Circulation 1999: 99: 2132-37.
46. Harris KM, Spirito P, Maron MS, Zenovich AG, Formisano F, Lesser JR, et al. Prevalence, clinical profile and significance of left ventricular remodeling in the end stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114: 216-25.
47. Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36: 2212-8.
48. McKenna WJ, Behr ER. Hypertrophic cardiomyopathy: management, risk stratification and prevention of sudden death. Heart 2002; 87: 169-76.
49. Frenneaux MP. Assessing de risk of sudden cardiac death in a patient with hypertrophic cardiomyopathy. Heart 2004; 90: 570-5.
50. Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation 2010; 121: 445-56.
51. Boriani G, Maron B J, Shen W-K, Spirito P. Prevention of sudden death in hypertrophic cardiomyopathy. ¿But which defibrillator for which patient? Circulation 2004; 110: e438-e442.
52. Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 2007; 298(4): 405-12.
53. Eckart RE, Scoville SL, Campbell CL, Shry EA, Stajduhar KC, Potter RN, et al. Sudden death in young adults: A 25 year review of autopsies in military recruits. Ann Intern Med 2004; 141: 829-34.
54. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin 2007; 25: 399-414.
55. Rowland T. Sudden unexpected death in young athletes: Reconsidering hypertrophic cardiomyopathy. Pediatrics 2009; 123: 1217-22.
56. Spirito P, Seidman CE, McKenna W, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336: 775-85.
57. Yetman AT, Hamilton RM, Benson LN, McCrindle BW. Long term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. J Am Coll Cardiol 1998; 32: 1943-50.
58. Savage DD, Seides SF, Clark CE, Henry WL, Maron BJ, Robinson FC, et al. Electrocardiographic findings in patients with obstructive and nonobstructive hypertrophic cardiomyopathy. Circulation 1978; 58: 402-8.
59. McLeod CJ, Ackerman MJ, Nishimura RA, Tajik AJ, Gersh BJ, Ommen SR. Outcome of patients with hypertrophic cardiomyopathy and a normal electrocardiogram. J Am Coll Cardiol 2009; 54: 229-33.
60. Sociedad Argentina de Cardiología. Miocardiopatía hipertrófica. Consenso Argentino SAC. Versión resumida. Rev Arg Cardiol 2009; 77: 1-28.
61. Charron P, Dubourg O, Desnos M, Isnard R, Hagege A, Millaire A, et al. Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in a genotyped adult population. Circulation 1997; 96: 214-9.
62. Charron P, Dubourg O, Desnos M, Bouhour JB, Isnard R, Hagege A, et al. Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in genotyped children. Eur Heart J 1998; 19: 1377-82.
63. Erice B, Romero C, Andériz M, Gorostiaga E, Izquierdo M, Ibáñez J. Erice B, Romero C, Andériz M, et al. Diagnostic value of different electrocardiographic voltage criteria for hypertrophic cardiomyopathy in young people. Scand J Med Sci Sports 2009; 19: 356-63.
64. Konno T, Shimizu M, Ino H, Yamaguchi M, Terai H, Uchiyama K, et al. Diagnostic value of abnormal Q waves for identification of preclinical carriers of hypertrophic cardiomyopathy based on a molecular genetic diagnosis. Eur Heart J 2004; 25: 246-51.
65. Afonso LC, Bernal J, Bax JJ, Abraham TP. Echocardiography in hypertrophic cardiomyopathy. The role of conventional and emerging technologies. J Am Coll Cardiol Img 2008; 1: 787-800.
66. Rickers C, Wilke NM, Jerosch-Herold M, Casey SA, Panse P, Panse N, et al. Utility of cardiac magnetic resonance imaging in the diagnostic of hypertrophic cardiomyopathy. Circulation 2005; 112: 855-61.
67. Maron MS, Maron BJ, Harrigan C, Buros J, Gibson CM, Olivotto I, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance., J Am Coll Cardiol 2009; 54: 220-8.
68. Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part 2. Differential diagnosis, risk stratification and post treatment MRI appearances. AJR 2007; 189: 1344-52.
69. O'Hanlon R, Grasso A, Roughton M, Moon JC, Clark S, Wage R, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56: 867-74.
70. Bruder O, Wagner A, Jensen CJ, Schneider S, Ong P, Kispert EM, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010: 56: 875-87.
71. Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114: 2232-9.
72. Ciampi Q, Betocchi S, Lombardi R, Manganelli F, Storto G, Losi MA, et al. Hemodynamic determinants of exercise induced abnormal blood pressure response in hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 40: 278-84.
73. Sen-Chowdhry S, McKenna W. Noninvasive risk stratification in hypertrophic cardiomyopathy: don’t throw out the baby with the bathwater. Eur Heart J 2008; 29: 1600-2.
74. Adabag AS, Casey SA, Kuskowski MA, Zenovich AG, Maron BJ. Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 45: 697-704.
75. Wang L, Seidman JG, Seidman CE. Narrative review: Harnessing molecular genetics for the diagnostic and management of hypertrophic cardiomyopathy. Ann Intern Med 2010; 152: 513-20.
76. Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol 2009; 54: 201-11.
77. Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 2008; 83: 630-8.
78. Landstrom AP, Ackerman MJ. Controversies in cardiovascular medicine. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation 2010; 122: 2441-50.
79. Olivotto I, Cecchi F. The epidemiologic evolution and present perception of hypertrophic cardiomyopathy. Ital Heart J 2003; 4: 596-601.
80. Maron BJ. Hypertrophic cardiomyopathy: An important global disease. Am J Med 2004; 116: 63-6.


{kind=link}
{kind=link}
{kind=link}